QMSR is here! Is your quality system ready?

Solutions built for medical device teams to move faster, stay compliant, and grow with confidence.
Manage quality events, training, and documentation in one place.
Control documents, training, and change with traceability.
Track CAPAs, audits, and NCs with audit-ready records.
Qualify suppliers and manage parts with visibility.
Connect design, risk, and AI-powered traceability as you build.
Link needs, requirements, and verification to design controls.
Automate traceability from dev tools for faster releases.
Assess ISO 14971 risk as requirements and tests evolve.
Oversee studies with connected data, documents, and workflows.
Capture compliant clinical data with a validated medical EDC.
Plan and manage PMCF aligned to EU MDR expectations.
Collect clinical outcome data in-person or remotely on any device.
Built with regulatory insight to help medtech teams move faster with confidence.
Find issues faster and make smarter decisions with less manual work.
Tools and resources that help teams implement quickly and meet requirements.


A QMS, or quality management system is a formalized system that documents the policies, procedures, documentation requirements, and processes that MedTech companies use to ensure that their products are both safe and effective for the end-user.
QMS stands for quality management system. A QMS is a documented framework that defines how an organization manages quality, controls processes, maintains records, meets regulatory requirements, and improves products or services over time.
For medical device companies, a QMS helps prove that products are designed, manufactured, monitored, and improved according to applicable regulatory standards and customer requirements. It defines how teams manage design controls, document control, risk management, supplier quality, corrective action and preventive action (CAPA), audits, training, complaints, and regulatory compliance. This guide explains QMS meaning, core QMS requirements, QMS examples, QMS vs eQMS, and how FDA QMSR and ISO 13485 shape medical device quality systems.
Organizations across multiple industries depend on their QMS to manage the quality, safety, and effectiveness of their product or service over a period of time, verifying and validating its ongoing performance and ability to meet the needs of customers requirements.
In the medical device industry, companies create their own policies, based on business needs, customer needs, the perceived risk of your device, specific requirements of your device, and compliance requirements of industry regulations. That information is stored in a QMS where the company manages its quality processes and activities that take place throughout the product lifecycle to ensure quality standards are consistently being met.
To better understand what a quality management system is, we need to break down the language that defines QMS. When someone refers to to their quality system, they are referring to the organizational structure, responsibilities, procedures, processes, and resources for implementing quality management.
Medical device QMS software is a digital QMS that helps MedTech teams run a Quality Management System in one place. It helps automate quality work (like document control, change control, training, CAPA, and audits) so you can stay compliant with standards like ISO 13485 and regulations like the FDA’s 21 CFR Part 820. It also helps companies reduce mistakes and keep quality data organized across the full device lifecycle.
Greenlight Guru is a purpose-built eQMS for medical device companies, with modules for document management, change management, CAPA & nonconformance, audit management, supplier management, and training management.
A QMS is used to store a company’s core set of business policies, procedures, forms, and work instructions, along with their sequence, interactions, and resources required to conduct business.
As you can see below, there are numerous items that must be addressed when implementing your QMS for medical devices.
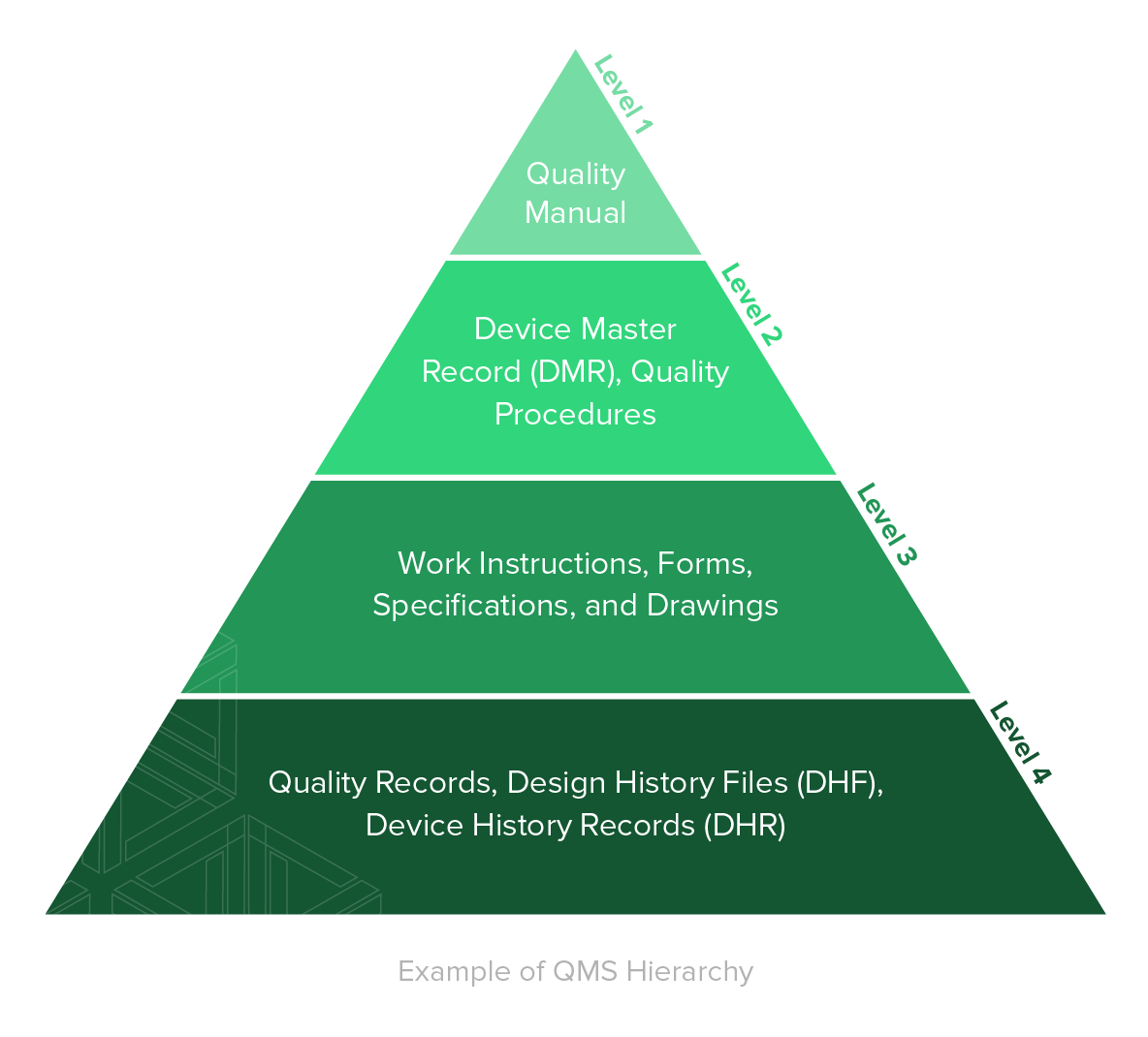
QMS Area |
Example |
| Document Control | SOPs, work instructions, forms, policies, version history, and approval records |
| Design controls | User needs, design inputs, design outputs, verification, validation, and design reviews |
| Risk management | Risk analysis, risk controls, risk acceptability criteria, and risk management files |
| CAPA | Corrective and preventive action records, investigations, root cause analysis, action plans, and effectiveness checks |
| Supplier quality |
Approved supplier lists, supplier qualification records, supplier audits, and supplier performance monitoring |
|
Training |
Role-based training assignments, training records, sign-offs, and retraining evidence |
|
Audits |
Internal audit plans, supplier audit findings, corrective actions, and audit reports |
|
Production controls |
Manufacturing instructions, acceptance criteria, inspection records, and process controls |
|
Complaints |
Complaint intake, investigation records, reportability decisions, and post-market trend data |
A practical medical device QMS software should include:
A QMS is more than just documentation, it's a set of functional, ongoing processes designed to consistently produce medical devices that are safe and effective for the end user.
Medical device companies that are required to establish a QMS to support product safety must appropriately structure oversight and accountability of such a system to ensure that the system is effective and that it is followed where required by all employees at the company. There are several QMS processes that fall under management controls, including management review of the quality system, internal quality audit procedures and the management approval process which applies to all documents in the QMS.
In a QMS, medical device companies must establish a set of processes known as design controls to help ensure that the medical device being designed and manufactured is safe and effective for the intended user, while accurately addressing user needs that reflect the design inputs and requirements. The design control process helps ensure that manufacturers not only make devices adequately, but that they also make the correct device that effectively addresses and satisfies the identified user needs.
The QMS is centered around the Corrective and Preventive Action (CAPA) process. As an organization collects feedback on its devices, it may become aware of product defects that are caused by errors in the manufacturing, storage or transportation of the medical device. When a nonconformance event occurs, medical device companies must maintain a process for taking corrective action to ensure that the event does not happen again. When a possible source of nonconformance is identified, a preventive action process can be initiated to help ensure that the issue is addressed before it leads to an adverse event.
A medical device QMS includes the documents, records, processes, responsibilities, and controls needed to manage product quality and regulatory compliance. While every company’s QMS should be right-sized to its device, risk profile, and business model, most medical device quality systems include the following elements:
QMS Element |
What It Includes |
| Quality manual or quality policy | The organization’s quality objectives, scope, responsibilities, and quality system structure |
| Standard operating procedures | Documented procedures that define how quality and regulatory processes are performed |
| Work instructions and forms | Detailed instructions and templates used to execute controlled processes |
| Quality records | Evidence that QMS procedures are being followed |
| Design and Development File | Records showing how a device was designed and developed |
| Medical Device File | Specifications, drawings, instructions, and other information needed to manufacture the device |
| Device History Record | Records showing that a device was manufactured according to the Medical Device File (now exists as part of the Production and Process controls under QMSR). |
| Training records | Evidence that employees are trained on relevant procedures and responsibilities |
| Supplier records | Supplier qualification, monitoring, audit, and performance documentation |
| Audit records | Internal audit, supplier audit, findings, and corrective action documentation |
A QMS improves day-to-day execution by turning “tribal knowledge” into repeatable standard operating procedures, then proving they’re followed with records and audit trails. It helps teams define what should happen, document that it happened, measure whether it worked, and improve the process when gaps appear.
A strong QMS supports:
Track performance metrics in your QMS like: CAPA cycle time, audit finding closure time, training completion rate, change-control lead time, complaint trend rate, and supplier defect rate (incoming defects per lot).
It used to be that medical device companies consisted of a quality manager and filing cabinet. Today, the QMS gives everyone – from quality, engineering, manufacturing, leadership, or other MedTech teams – a single source of truth to manage the quality of their product.
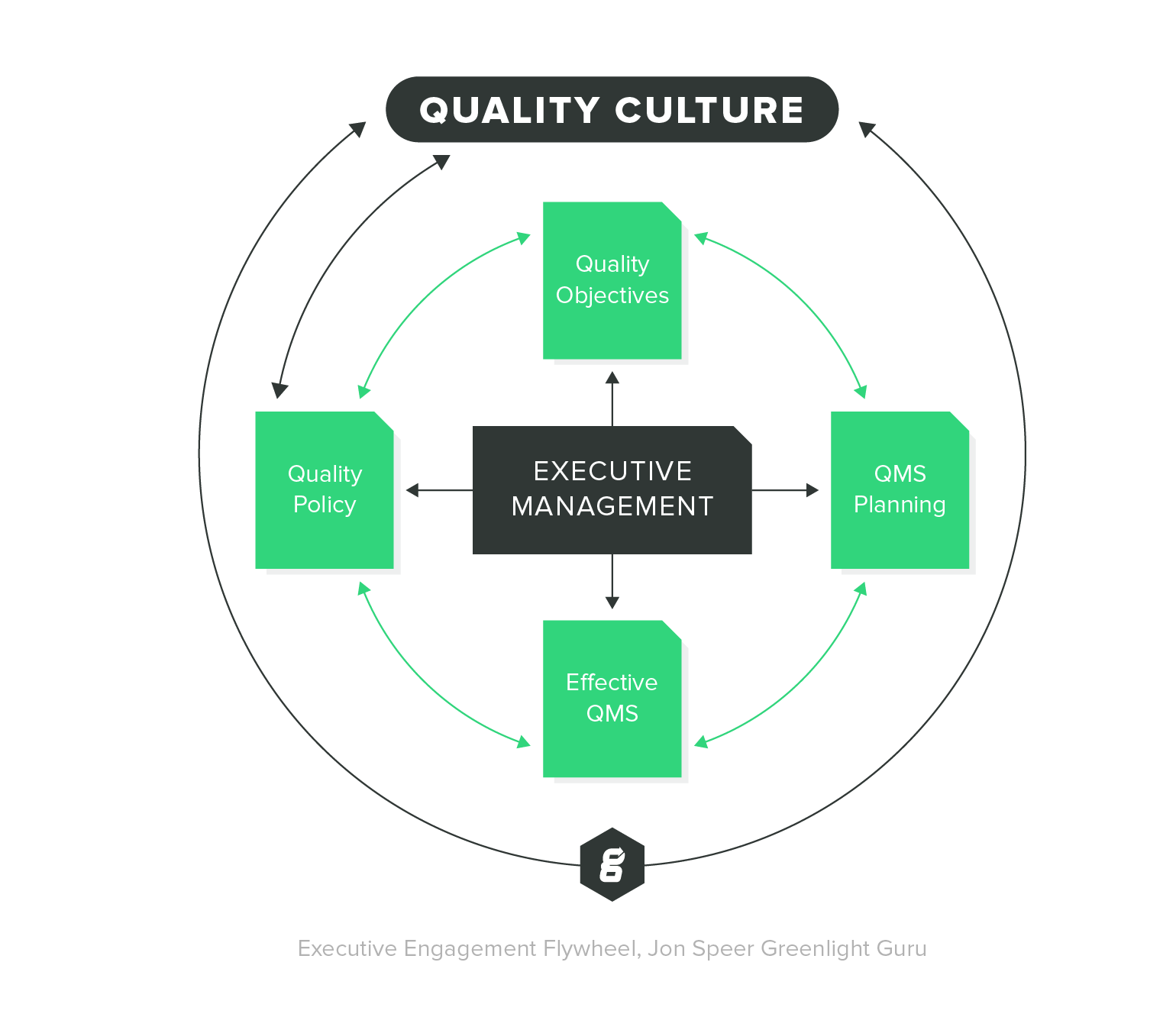
A QMS tool lets you store key quality documents and artifacts, track corrective and preventive actions (CAPAs), promote effective product development, continually monitor process improvement, and adhere to industry standards and regulations - all in one central location to make information available to anyone at your organization who might need it.
With visibility and easy access to quality records and data, it’s easier to collaborate and make quality a shared responsibility throughout your organization. Everyone in your company can see the documented controls for supplier management, production processes, labeling, packaging, handling, and even storage of your product, so you can maintain complete centralized control over the products that you create.
In order to promote standardized approaches to total quality management, regulatory bodies around the world enforce QMS requirements. These vary by geographic region with each market having a designated regulatory authority in charge of enforcing QMS regulations and standards.
There are several regulations and standards with specific QMS requirements for medical devices:
In the United States, a QMS for medical devices is regulated by the Food and Drug Administration (FDA). FDA provides a framework of basic, non-prescriptive requirements for manufacturers in 21 CFR Part 820, the Quality System Regulation (QSR) for medical devices.
According to FDA, medical device manufacturers are required to develop a QMS that is consistent with the:
It is, however, ultimately up to the manufacturer to assign roles and responsibilities of the persons who will be executing such processes using a risk-based approach. Risk mitigation and reduction methods, when done effectively, can have a strong positive impact on both the customer satisfaction and the business.
FDA QMS requirements state that the manufacturer is responsible for creating their organization’s policies based on meeting the customer requirements, perceived product risk, and requirements specific to the device and applicable regulations and standards.
Medical device companies that implement electronic records or electronic signatures as part of their FDA-mandated QMS must look to 21 CFR Part 11 for the specific guidelines that permit the use of technology in quality systems.
QMS software has changed the way that the FDA thinks about record keeping and electronic signatures as they pertain to quality systems. Your quality manager is frequently required to sign, date, and approve certain documents generated as part of your QMS.
However, if your entire QMS is electronic, what controls are in place to ensure that the electronic signatures provided have the exact same weight and meaning as a physical signature? The controls which permit the use of electronic signatures and records as part of a QMS are outlined in 21 CFR Part 11.
ISO 13485:2016 is the internationally-recognized standard for QMS for medical devices. Published by the International Organization for Standardization (ISO), the globally harmonized standard specifies requirements for medical device manufacturers to obtain certification of their QMS in accordance with customers and the applicable regulatory requirements.
QMS requirements under ISO 13485:2016 apply to every stage throughout the entire lifecycle of a medical device and can be applied by medical device manufacturers, suppliers or any other third-party entity that provides QMS-related services to the primary medical device organization.
To comply with ISO 13485 QMS requirements, manufacturers must ensure all contents within the QMS align with applicable requirements of the standard and those specific to the market in which your medical device is manufactured and sold.
ISO 9001 is part of the larger ISO 9000 family of standards, all of which are concerned with particular aspects of quality management. This family of standards is generic, which means they can be used by companies of any size, in any industry, to implement a QMS.
ISO 9001:2015 is the best-known standard in the ISO 9000 family because it lays out the basic requirements for implementing the processes, procedures, and documentation that makes up a QMS. If you’re a medical device manufacturer, however, there’s really no reason to follow ISO 9001 instead of a more MedTech-specific ISO standard such as ISO 13485.
An eQMS (electronic quality management system) replaces existing, outdated versions of QMS tools that often consist of physical paper stored in physical storage locations, like filing cabinets and desk drawers.
QMS software eliminates the cumbersome nature of non-electronic QMS tools by providing a single source of truth for organizations to manage the quality of their product or service.
A QMS platform for medical devices is an industry-specific solution that’s designed from the ground up to address and cater to the specific needs of the MedTech industry. These purpose-built QMS solutions often come with regulatory compliance protocols built into the system that are specific to the niche vertical market of medical devices.
The best QMS software allows medical device manufacturers to easily keep up-to-date records, makes documents easily accessible to all key stakeholders, and has dedicated workflows built-in to the system that align with industry standards and regulations.
Purpose-built QMS software can be a major boon for your medical device business, in several key areas.
The corrective and preventive action (CAPA) process is the heart of an effective QMS. This set of policies and procedures governs how an organization will assess feedback about a nonconforming product and use it to correct sources of nonconformance in its product design, supply chain, and manufacturing processes.
CAPA can generate a high volume of documentation, but Greenlight Guru's QMS software helps you stay organized with assigned case numbers for each CAPA request and a streamlined approvals and review process.
The goal of an effective product development team is to create a product that meets identified user needs and is both safe and effective for its intended use. Medical device companies implement design controls, a crucial set of QMS processes, to ensure that product development follows the needs of the user without getting off track.
Greenlight Guru’s QMS software helps by providing a single platform where your engineers can securely store and access all of the documentation produced in the product development process.
With a software-based QMS, you'll never misplace or lose track of the most important documents related to your device.
Greenlight Guru's QMS software allows anyone at your organization to find and reference the policies most relevant to their role. By establishing and documenting controls for supplier management, production processes, labeling, packaging, handling, and even storage of your product, you maintain complete centralized control over the products that you create.
QMS software solutions built with cloud-based technology also provide:
Manufacturers are responsible for developing, controlling and monitoring processes to ensure that manufactured medical devices conform to the required specifications. There are also quality management processes used to test products for conformance once the manufacturing process has been completed.
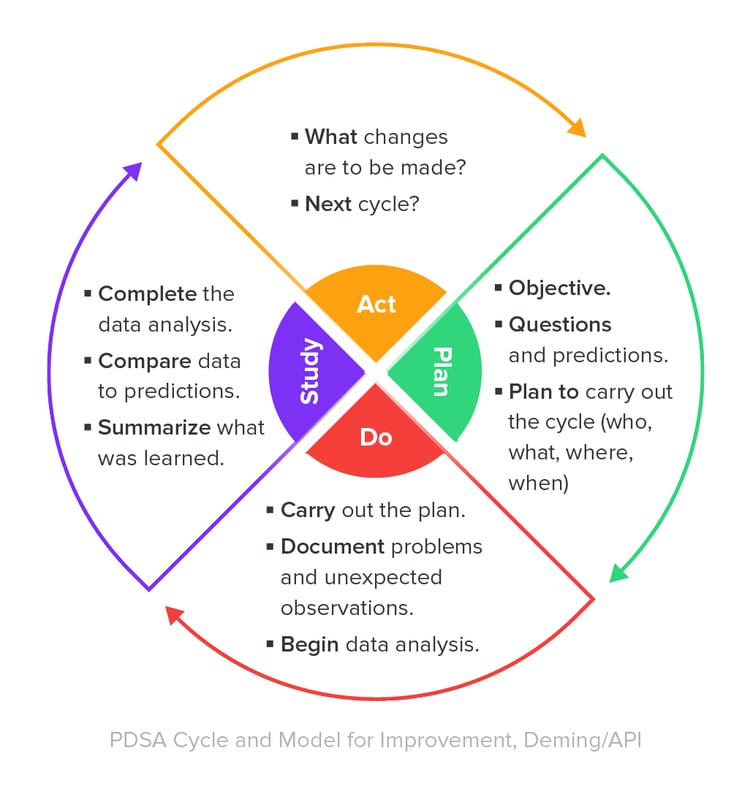
A valuable best practice for managing a QMS is to continually monitor its effectiveness and ensure that the QMS is adjusted as necessary.
One means to do so is to establish key performance indicators for the processes within the QMS. Consider applying a “Deming Cycle” methodology for your QMS effectiveness monitoring.



.webp?length=374&name=CaseStudy-200x200-01%20(1).webp?format=webp)







