.png?width=210&name=ISO_TOC_0%20(1).png)
.png)













Table of Contents
What is ISO 13485?
ISO 13485 is the global standard for medical device quality management systems established by the international standards organization (ISO).How do you manage your Quality Management System? If you are like the majority of the medical device industry, chances are you have a QMS that is a combination of paper-based processes and general-purpose tools, loosely held together by a group of people within your company--usually document control.
It’s important because it is long overdue with the previous version being released 13 years earlier in 2003.
ISO 13485:2016 is very much a bridge. What I mean is that this bridge explicitly describes and defines current QMS expectations for medical device companies. Prior to these refinements being formally defined and documented in the standard, many of the best practices being advised and adopted were very ad hoc in nature and often seemed to be based on auditor opinions.
Traceability
The best way to describe this approach to a QMS is ad hoc. I mean no disrespect if this is your method. It can work. But it is full of risks. Risks of relying on tools that don’t scale. Risks of inefficiencies. Risks that QMS knowledge lies solely with the people managing the day-to-day.
Some medical device companies have progressed from ad hoc to eQMS software tools that are highly customizable and configurable. While on one hand, this approach seems more advanced, this approach also comes with risks. Anytime a tool is configurable, what assurances do you have that it aligns with the applicable regulations and requirements—in this case, ISO 13485:2016? And how do you validate this?
It’s because of these reasons and my own personal experiences with these QMS approaches that led me to start Greenlight Guru. Our team of medical device industry experts has designed and built an eQMS software platform specifically for the medical device industry. Our industry-specific platform is architected to support the requirements of ISO 13485:2016 (and other regulatory requirements), where they are addressed automatically with no configuration required.
Greenlight Guru is a purpose-built solution for the medical device industry that addresses compliance, allowing you to better focus on developing high-quality products through streamlined processes.
Bottom line: This approach allows you to put emphasis on what is best for the patients who receive your life-saving technologies.
Note: If you would like to schedule a demo of Greenlight Guru's Quality Management Software, click here »
Evolution of a Quality Management System
Many view a Quality Management System (QMS) as a necessary evil for a medical device company—something you must have in order to be compliant.
A QMS is often times seen as the set of procedures that define the rules and restrictions that must be followed in the quest for designing, developing, and manufacturing medical devices.
Few embrace the notion that a QMS is beneficial. And frankly, most companies implement a QMS that is largely rule-based, restrictive, cumbersome, and largely ineffective.
The conventional approach for establishing a medical device quality management system is that of addressing compliance to regulations—sometimes resulting in direct regurgitation of requirements defined in ISO 13485:2016 and other quality system requirements and regulations.
If you see no issues with this approach and your opinion is that a QMS is simply a means to demonstrate regulatory compliance, then this guide may not be for you.
My view is that a QMS should be a set of processes that help me to run a better, more efficient business that focuses on true quality and what is best for the patients who will be recipients of my medical devices.
Let me take a moment to get us on the same page regarding Quality and a Quality Management System.
For me, when I hear the word “quality”, two names come to mind: W. Edwards Deming and Joseph Juran. Both Deming and Juran made significant contributions to quality and QMS as we know it. Juran was especially focused on quality management and is often regarded as the “father of modern quality management.”
It should be noted that the principles of quality from Deming, Juran, and a few other founding fathers of modern quality were established decades ago. In fact, Juran wrote about the “cost of poor quality” or the costs associated with providing poor quality products and services, back in 1951.
So why is the medical device industry still struggling with quality and quality management? Why do these principles still seem new and novel?
Now consider your QMS as the story of your business. How you function. How you operate. The story of how your company designs and manufactures medical devices. The story of how your company addresses product and process issues. The story of how you ensure product and process quality is essential and part of your core. And how patient safety and product efficacy matter.
You may have this desire to tell a compelling story with your QMS and wonder how you can achieve this. This guide will help provide insights, tips, and pointers on how to do so.
Before diving in too deep into this ISO 13485 guide, I think it is beneficial to understand a bit of history as to how the current industry view of QMS came to be. Without going through the entire evolution of quality system regulations and requirements, let me instead share a fairly typical story of how many companies established their QMS and why the medical device industry struggles with this.
At some point in time, a company understood or realized that there were certain compliance needs to address in order to be a medical device company. The decision to define processes and procedures comprising a quality management system was more or less a decision of a “we have to,” rather than a “we get to.” To say it another way, companies implemented a QMS because they were more or less forced to comply with applicable regulations governing medical device companies.
Over time, as audits and inspections happened, the QMS processes and procedures were edited, often times to satisfy the request of an auditor. And as a result, new procedures were defined. The company would add new people, sometimes growing, sometimes outsourcing. As a result, existing procedures would be edited and/or more new procedures defined.
The distance and operational interactions between functional groups grew greater and wider. The quality group was often viewed as the people who made all the rules and restrictions that slowed the company down. And this view isolated quality—not a thing we should embrace, but more as something to resist.
This cycle repeated over and over. All the while, the company’s QMS was being shaped and shifted in an attempt to meet the changing company and compliance needs—becoming more and more unwieldy, dysfunctional, and burdensome. The company’s QMS was causing the business to slow down. Or worse, employees were finding workarounds or ignoring QMS procedures altogether. The notion of quality became viewed as a function of compliance and an impediment.
With all of that said, it’s now time to provide you with a guide to ISO 13485.
A guide that bridges meeting requirements of ISO 13485:2016 in a way to help your business rediscover (or discover for the first time) how true quality should be the guiding force to improve your products and processes in a way that puts patients first. Yes, meeting requirements and being compliant are important. Yet, this should not be the end-goal and primary objective for your QMS—true quality is, or at least should be.
When you shift your focus to what is best for the patient, then compliance becomes a natural by-product.
Should you follow the requirements defined in ISO 13485:2016 and become certified? Technically, no you don’t have to. Will doing so help you run a better business where patients will receive benefits? Absolutely.
The rest of this guide will, in large part, follow the major sections and headings of ISO 13485:2016 providing you specific, actionable steps and best practices you can apply at your medical device company. My goal is to make sure your company not only complies with ISO 13485:2016, but also allow your company to be focused on true quality.
.png)
Requirements for ISO 13485 Certification
I view the establishment of ISO 13485:2016 standard as an important milestone for the medical device industry.
It’s important because it is long overdue with the previous version being released 13 years earlier in 2003.
The 2016 standard is very much a bridge. What I mean is that this bridge explicitly describes and defines current QMS expectations for medical device companies. Prior to these refinements being formally defined and documented in the standard, many of the best practices being advised and adopted were very ad hoc in nature and often seemed to be based on auditor opinions.
What is a Quality Management System?
According to ASQ, a quality management system is defined as:
“A formal system that documents the structure, processes, roles, responsibilities and procedures required to achieve effective quality management.”
A QMS is comprised of the core set of business policies, procedures, forms, and work instructions, along with their sequence, interactions, and resources required to conduct business within a medical device company. Quality records are documentation that demonstrate the QMS is being executed and followed.
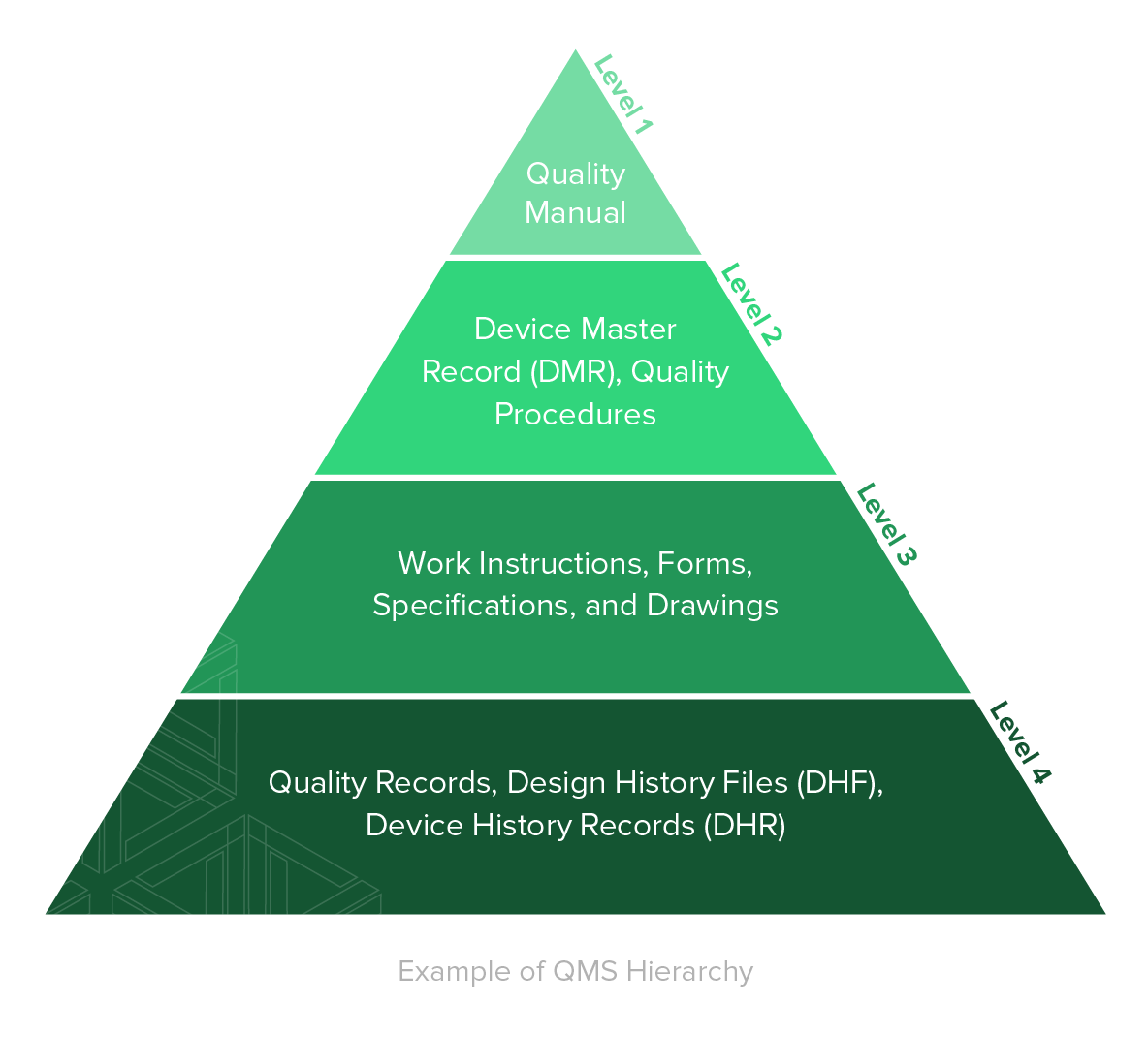
To align with ISO 13485:2016, contents of the QMS should address the specific, applicable requirements of the standard, as well as the specific, applicable regulatory requirements based on markets where you plan to manufacture and market medical devices.
One concept formally introduced in the 2016 standard is the notion of a risk-based QMS. Throughout this guide, I will revisit and emphasize what “risk-based” means and how it applies to the various aspects of a QMS.
If you choose to outsource any process(es) that impacts requirements of the standard (for example, contract manufacturing), it is your responsibility to monitor and ensure controls over the outsourced processes. This includes defining roles and responsibilities in documented quality agreements with any outsourcing resources.
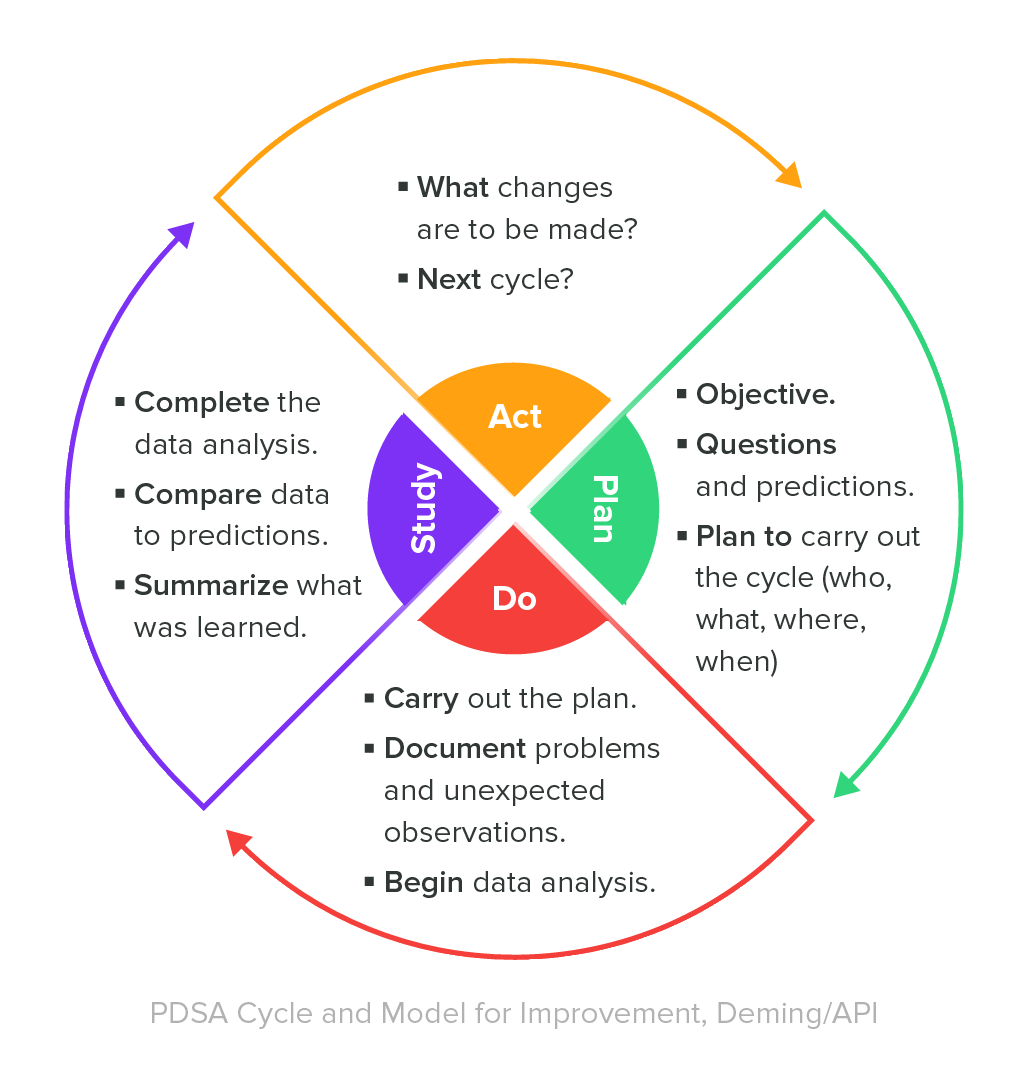
A valuable best practice for managing a QMS is to continually monitor its effectiveness and ensure that the QMS is adjusted as necessary. One means to do so is to establish key performance indicators for the processes within the QMS. Consider applying a “Deming Cycle” methodology for your QMS effectiveness monitoring.
In the event you use an eQMS software tool, software validation procedures must be defined and the system must be validated before use.
It’s worth noting that validating most eQMS tools available to you will be time consuming and frustrating. Why? Most eQMS tools are general purpose and not specifically aligned to ISO 13485:2016 and medical device QMS requirements.
This means the first step you have to do to even use these types of tools is to customize and configure to align with ISO 13485. And this easily takes several months. Then, once configured, you have to define protocols and execute testing in order to validate the tool--again taking weeks and months to complete.
At Greenlight Guru, it’s our goal to alleviate those efforts and streamline your processes through our multi-tenant, cloud-based SaaS platform. According to a LNS Research ‘State of the Market’ piece on Software Validation in the Life Sciences industry, they assert,
Cloud-based technologies create new opportunities to streamline validation [and] industry-leading vendors are providing pre-validated platforms, pre-validated functions, and pre-validated pre-configurations.
This notion of streamlined processes is commensurate with the “no-effort” approach, a method we’ve pioneered with our software. By establishing an automated system for the validation process, companies are given a steadfast track for compliance with 21 CFR Part 11.
![ISO-13485-documentation-requirements]() ISO 13485 Documentation Requirements
ISO 13485 Documentation Requirements
By now, you should be very familiar with the adage “...if it isn’t documented, then it didn’t happen.”
Yes, documentation of QMS processes, quality events, and workflows is critically important. Sometimes the notion of documentation can create angst within a company. Sometimes the idea of documentation is viewed as overly burdensome and often times unnecessary with little value added.
In my experience, most companies do create quite a few burdens and obstacles regarding documentation. And it doesn’t have to be this way.
Documentation should be about defining processes and maintaining the records required to demonstrate these processes are being followed. Documentation is key for objective evidence. Objective evidence to support your employees through design, development, manufacturing, and support of medical devices. Objective evidence to demonstrate that requirements are being addressed.
Establishing thorough, yet functional, document management practices for your business is one of the most important foundational elements of a QMS.
1. Quality Manual
A key part of your medical device quality management system is your quality manual.
The common approach for satisfying the guidelines of ISO 13485:2016 for a quality manual is creating a lengthy policy-level document that breaks down various sections of the standard and describes from a high-level how the medical device company addresses the clauses. This approach is fine.
Your quality manual must meet the following criteria:
- Describe the scope of your QMS. Include any clauses that are excluded or non-applications supported with justification.
- List or reference the procedures comprising the QMS.
- Describe interactions of QMS processes.
- Outline the structure of QMS documentation.
2. Medical Device File
Every medical device type or device family must have a medical device file.
ISO 13485 requires the contents of a medical device file to include:
- Description of the product, including intended use and indications for use.
- Product labeling and instructions for use.
- Specifications for the product.
- Specifications and procedures for manufacturing, inspection, labeling, packaging, storage, handling, and distribution.
- Specifications for measuring and monitoring.
- Specifications and procedures for product installation (if applicable).
- Procedures for product servicing (if applicable).
3. Document Control
Documentation is a necessary aspect of a quality management system.
A document control procedure shall define your company’s criteria for document control. This includes ensuring documents are reviewed and approved prior to implementation, means to revise documents and identify changes, and ensure current versions are available at point of use.
4. Control of Records
Records require similar controls as documents. And sometimes the distinction between a document and record can be a bit confusing.
Records are evidence that certain processes have been followed. Throughout this guide, there are additional notes related to records.
The same type of criteria is applicable regarding review and approval. Records, however, are typically not versioned.
![ISO 13485 management responsibility]() Management Responsibility
Management Responsibility
I’ve experienced and observed many companies where the employees embraced a true quality mindset that was not supported by executive management.
Some in these situations believed that quality could be a grassroots type of campaign and that management would eventually come around. Unfortunately, it seldom works out this way.
Executive management sets the tone with respect to your company’s vision and importance of quality. For a QMS to be effective, executive management within a medical device company has to believe in its importance. Both Deming and Juran emphasized this.
“Management’s job is to know which systems are stable and which are not.” - Deming
“It is most important that top management be quality-minded. In the absence of sincere manifestation of interest at the top, little will happen below.” - Juran
And executive management has to do more than just pay lip service to the QMS. They have to support it, embrace it, and live it. True quality has to be part of the culture. The moment executive management slips in their approach to true quality, it becomes difficult to reestablish.
A significant part of a healthy quality culture focuses on the customers of the company. For medical device companies, the ultimate customer is the patient receiving the devices and technologies designed, developed, and manufactured. Always doing what is best for the patient becomes the guiding force for true quality.
Quality Policy
The quality policy according to ISO 13485 is the keystone to ensuring effectiveness of your quality management system.
Okay, I can imagine what you might be thinking. Yes, many companies create a quality policy statement that seems full of altruistic and fluffy words. And for many companies, the quality policy is plastered on posters hung on walls throughout the office and occasionally distributed to employees on laminated cards. A quality policy statement often feels very cliché, at best.
The quality policy of your company should reflect management’s commitment to quality. It should express the ultimate purpose of your organization. It should be communicated and understood throughout the company. It should express the culture of quality of the company and be something that is believed at all levels of the organization.
Quality Management System Planning
Planning is an important element in order to ensure a QMS is effective. And even if you already have a QMS that is implemented, audited, and certified, there are benefits to initiating QMS planning efforts.
QMS planning should incorporate identification of key quality objectives for the company. Think of quality objectives like goals; these should be objective and measurable. And quality objectives should flow from your quality policy.
Regulations evolve, your company introduces new products, new markets are added, processes change, products change, and so on. All reasons to embrace the idea of QMS planning with defined quality objectives.
Responsibility, Authority & Communication
As I previously stressed, executive management has the ultimate responsibility of ensuring your QMS is effective. As part of this, management needs to ensure that roles and responsibilities of the QMS are well defined.
This includes providing the necessary authority to applicable personnel regarding QMS initiatives. Executive management needs to appoint a management representative. The management representative must have the authority to oversee and manage your company’s QMS efforts.
Management Review
It is expected and required that a medical device company establish processes for conducting planned management reviews. In my experience, most companies do this annually, and unfortunately, it often seems like a checkbox type of activity to say it was done.
I hope by now, though, you are noticing a theme. The theme is executive management establishes and embraces your company’s quality culture, the quality policy aligns with the quality culture, quality objectives are defined that are consistent with quality policy, and QMS planning ensures quality objectives are addressed and QMS is effective.

Management review is a continuation of this theme. It is a moment in time to ensure QMS initiatives are on track and in alignment with the company’s quality policy.
In addition to reviewing the quality policy, quality objectives, and QMS planning, management review should also evaluate:
- Customer feedback and complaints
- Adverse events and regulatory reporting
- Internal, external, and supplier audits
- KPIs for processes, including any non-conformances
- KPIs for products
- Status of corrective actions
- Status of preventive actions
- Follow-ups from previous management reviews
- Issues that could impact the QMS, including updates to regulatory requirements
- Opportunities for improvement.
The ultimate objective of management review is to evaluate suitability, adequacy, and effectiveness of your QMS.
As I mentioned, the norm is to conduct management review annually, or maybe twice per year. Preparing for management review is very time-consuming. Often times, preparing the data and information regarding various products and processes requires the quality manager and other key resources living in spreadsheets for hours--sometimes days--just to prepare for management review.
Part of the reason for this is because most companies are dealing with an unwieldy (and frankly ineffective) QMS where data and information is buried in various files and locations. The necessary information is not collected in a single source of truth.
Is this why management reviews are largely checkbox activities? Is this why companies conduct management reviews once per year?
My recommendation is to hold management reviews at least once per quarter. I realize that in order to do so, you need a QMS that is more intuitive, user-friendly, and provides access to data and information more easily.
ISO 13485 Resource Management
Your resources are important and critical for effectively running your business. According to ISO 13485, all companies must have the resources they need to complete their tasks effectively.
This includes the people, infrastructure, processes, and overall work environment. With respect to the QMS, having the appropriate resources is just as important. Regardless of how you deploy and implement your QMS, having the right people with the applicable training and experience is essential to helping ensure QMS effectiveness.
This premise emphasizes the importance of competency and training. And if you think about it, it totally makes sense. Do you want an employee to perform just any activity within your business without being qualified or properly trained?
Defining overall personnel training needs across your business are necessary. Furthermore, documenting training plans and maintaining objective evidence of employee training is good practice for your internal operations.
Another aspect of resource management includes buildings, workspaces, process equipment, software to support business operations, and support services. Your infrastructure must be appropriate for the types of activities and operations conducted by your business. For some types of products and processes, this may entail definition of cleanliness, PPE, and other environmental controls.
ISO 13485 Product Realization Planning
Product realization describes how your business designs, develops, manufactures, and delivers medical devices.
The guidelines in ISO 13485:2016 for product realization includes resources and processes required for defining customer needs, design and development, purchasing, production, and field support. From a high-level, product realization includes:
- Planning of Product Realization
- Customer-Related Processes
- Design and Development
- Purchasing
- Production and Service Provision
- Control of Monitoring and Measuring Equipment
Product realization involves several elements of your QMS and a variety of personnel and resources. It is important to appropriately plan product realization efforts to address the following:
- Quality criteria is established for the products.
- Required processes and supporting documentation are defined.
- Establish the appropriate infrastructure and work environment.
- Ensure personnel involved are competent and adequately trained.
- Verification, validation, monitoring, measurement, inspection, handling, storage, distribution, and traceability activities are established and commensurate with the products and processes.
- Documented records to demonstrate product realization processes and product meet defined requirements.
![ISO 13485 customer related processes]() ISO 13485 Customer-Related Processes
ISO 13485 Customer-Related Processes
Before you design and develop products, it is critical that you understand what needs and requirements are important to and required by customers.
Note, that the term “customer” is somewhat ambiguous. The usual, obvious customers are the patient and end-user. I urge you to consider other customers beyond just the patient and end-user for your product too. This can include regulatory agencies, distributors, purchasing organizations, and shipping and logistics resources.
A good way to think about who your customers are for your products is to consider who will interact with the product once it leaves your control--even if the product is packaged in a box in transit.
For example, if your product contains lithium-ion batteries, there are customer requirements that might pertain to your logistics and/or shipping resource to consider for how you label your product shipper.
Customer needs and requirements must be defined and documented. Consider user interactions and training that might be applicable. Consider regulatory requirements too.
Also don’t expect your customers to voluntarily articulate and communicate their needs and requirements to you. There’s a good chance you will have to extract this critical information from the various customer types for your products. And once defined, this information will be invaluable to your design and development processes.
The customer needs and requirements are also helpful for honing in on your product’s intended use and indications for use, which will support your overall efforts throughout design and development and regulatory submissions.
It surprises me how often customer needs and requirements are not well-defined and documented by companies. Failing to do so increases your business risk. Think about it. If you do a poor job of defining customer requirements, how do you know that you are designing and developing the right products?
Consider the alternative. Having well-defined customer requirements is invaluable information for your entire product realization efforts. My advice is to be risk-based and actually risk-averse by emphasizing the importance of customer needs and requirements throughout your product realization process.
One crucial piece of advice I’d like you to consider when documenting customer needs and requirements is to start constructing a design and development traceability matrix. Essentially, a well-constructed traceability matrix will show the flow of information from customer requirements throughout the design and development process.
The most common approach for implementing a traceability matrix is to use a spreadsheet. While this can be helpful for showing the relationships of design and development activities, keeping a traceability spreadsheet up to date throughout design and development and product realization will require hundreds of hours of time per project per year.
With Greenlight Guru, we have constructed a workflow within our eQMS software to easily capture and document customer requirements and other design and development items. This functionality also streamlines the traceability process, giving you back valuable time to focus on more value-add tasks and activities.
I’ll touch on design and development traceability a few more times below.
Another aspect to consider regarding customer-related processes is communication--internal and external. Be sure to communicate product information to your customers. Also establish processes for handling product questions, inquiries, ordering, feedback, complaints, and advisory notices.
ISO 13485 Design and Development processes
The design and development for ISO 13485 should be a continuation of customer needs and requirements.
Could you enter into design and development without documented customer needs and requirements? Theoretically, yes, you can. Is it a good idea? In my expert opinion: Doing so would be a terrible idea and incur significant business risks that can be largely avoided. I stress this because:
- Way too many design and development teams dive into product development without a clear understanding what is important about the eventual product from the perspective of the customers.
- Why are you developing medical devices to begin with? I assume it is for customers (patients). I assume it is to improve quality of life.
- Not having clarity on customer needs will more than likely result in costly reworks, revisions, and loss of time.
Do yourself a huge favor and define customer needs and requirements as comprehensively as possible.
With that said, let me offer one quick note. You do not need to have a complete list of customer needs to be documented before diving into design and development. It is perfectly acceptable to apply agile methodologies and be iterative. Just try to have enough clarity about what issue you are trying to solve before diving too deep into development.
Design and development processes include:
- Design and Development Planning
- Design and Development Inputs
- Design and Development Outputs
- Design and Development Reviews
- Design and Development Verification
- Design and Development Validation
- Design and Development Transfer
- Control of Design and Development Changes
- Design and Development Files
Keep in mind that design and development, as defined within ISO 13485:2016, is very much in sync with FDA design controls regulations defined in 21 CFR Part 820.30.
Let me share a few key tips and pointers regarding design and development here. For a complete, in-depth guide on design controls, let me refer you to The Ultimate Guide to Design Controls—the medical device industry’s #1 resource for design and development.
Design and Development Planning
Design and development planning is about understanding the scope of product development efforts.
Effective planning identifies applicable development phases or sprints, depending on the type of product development methodology used. Planning should include definition of the key deliverables of each phase/sprint. Planning should also define when during the development cycle you plan to conduct design and development reviews.
Another key aspect of design and development planning is to define the authorities and their responsibilities, who will be managing the process. You also need to identify any and all resources required and ensure those resources have the necessary competency and expertise. Planning should also describe verification, validation, and design transfer, as well as define how traceability of design and development activities will be addressed.
Some folks view design and development planning as an activity that is conducted towards the onset of a project only. If you use that approach, be aware that it is not a best practice.
Yes, planning is likely more intensive towards the beginning of the project. That said, design and development best practices embrace the notion that planning is an ongoing activity throughout the entire product development lifecycle. And at each subsequent phase/sprint, you need to revisit planning efforts and make adjustments.
Here’s why: there is no way that you can effectively define a plan for the entire product development process when just starting a project. As development occurs, you learn more, things change, and you have to make adjustments accordingly. The design and development plan needs to be very fluid.
Think about it this way. How well can you plan product development activities that are more than a few months out? Based on my experience, I can effectively plan the next phase/sprint. Trying to establish detailed design and development planning beyond this stage will likely be inaccurate and not useful. And planning should be a useful mechanism to communicate to the project team and company stakeholders about the product development project.
With a little bit of extra effort, you can incorporate your product risk management planning as part of design and development planning. I definitely recommend doing so because, in my opinion, risk management and design and development should flow as one process.
To address risk management planning in alignment with ISO 14971, there are some similar criteria as per design and development planning:
- Define scope of risk management activities; describe the medical device and its intended use.
- Identify life-cycle phases included.
- Assign roles, responsibilities, and authorities.
- Define risk management reviews.
- Establish criteria for risk acceptability.
- Determine risk verification activities.
- Describe activities pertaining to production and post-production risk information.
Like design and development planning, risk management planning should also be living throughout product realization.
Design and Development Inputs
Design and development inputs are where you capture and document all of the product requirements for the device being developed.
This includes functional, performance, usability, safety, regulatory, and applicable standards requirements based on the defined intended use.
Earlier, I talked about the importance of defining customer needs and requirements (sometimes these are referred to as “user needs”). Your customer requirements are important to understand so that you can properly define the design and development inputs.
A risk management process needs to be incorporated as part of design and development, as well as the entire product realization process. And defining your product requirements goes hand in hand with product risk management.
At this stage of development, you should start to understand the potential hazards, hazardous situations, and harms that could result based on product requirements and design decisions.
Essentially, you should be documenting your initial risk analysis in conjunction with defining customer needs and requirements and design and development inputs. Doing so will help you understand product risks and improve the overall product design.
The ISO 14971 standard is a terrific resource and includes quite a bit of helpful information regarding safety requirements for your product (refer to informative Annex C and Annex E in ISO 14971).
As you define design and development inputs, ensure that you are assessing that the product requirements are complete, unambiguous, verifiable, and not conflicting with one another.
Design and development inputs must be reviewed and approved by appropriate resources for accuracy and completeness. A good mechanism to do so is via design and development review.
Design and Development Outputs
As you progress from design and development inputs to defining specific details and characteristics about the product, you should be establishing drawings, specifications, manufacturing instructions, code, inspection procedures, purchasing specifications, and so on for the eventual medical device.
These items are your design and development outputs.
Outputs describe all the components, parts, pieces, assembly, and inspection elements required to purchase, manufacture, and service the medical device. I think of this as the “recipe” for your medical device--where you lay out the ingredients and steps involved to get the product to end users. This stage in the process occurs when you begin to establish the initial medical device file (described earlier in this guide).
Your design and development outputs should also:
- Contain or make reference to acceptance criteria.
- Identify essential characteristics to ensure product safety.
- Include your product labeling, instructions for use, and product packaging.
- Suitable to verify design and development inputs have been met.
Design and development outputs can be used as means to establish risk mitigations and risk control measures too. According to ISO 14971, risk control options should be prioritized by:
- Inherent safety by design.
- Protective measures in the medical device itself or in the manufacturing process.
- Information for safety.
Always capture the details of your design and development outputs as part of your product risk assessment.
Design and development outputs must be approved prior to release; this is part of the process of progressing towards the design and development transfer, in preparation for eventual manufacturing. And once again, update your traceability matrix to show how design and development outputs relate to inputs.
Again, a good way to do so is via a design and development review. I highly recommend releasing design and development outputs before conducting builds for design and development verification, animal studies, and clinical investigation.
Design and Development Reviews
I talked briefly about design and development reviews during planning, inputs, and outputs. Now let me expand a bit more on that subject.
The timing of design and development reviews should be in sync with design and development planning. During planning, you identify the stages during product development when design reviews are necessary. And as your product development progresses and project evolves, you should be revisiting the timing of these reviews.
The primary purpose of design and development reviews is to ensure that the product you are developing is able to meet customer needs and requirements and product requirements.
Reviews should include personnel, team members, and resources pertinent to the stage being reviewed in order to make this assessment. My advice is to also always include risk management information as part of your design and development reviews.
Design and Development Verification
Design and development verification is about demonstrating that you have designed your medical device correctly. In order to do so, you must conduct some type of test, inspection, or analysis to prove your design and development outputs meet your design and development inputs.
Verification requires that the plans, methods, and acceptance criteria be defined before executing the activities. In some types of verification activities, determining sample size according to accepted statistical techniques will be important. I recommend using established standards to help you with this part. Keep in mind that building product to design and development verification activities is also a part of the design and development transfer efforts.
Also, while not medical device specific and more general manufacturing product quality, I suggest reviewing “The Importers Guide Managing Product Quality with AQL (ebook)” for a thorough overview of the benefits of statistical techniques and sampling.
Upon completion of verification activities, results should be documented and demonstrate that outputs meet inputs. A common tool to assist with this is a traceability matrix. Design and development verifications should also be used as a means to demonstrate effectiveness of risk controls and captured as part of your product risk assessment.
Once again, a design and development review is a way to review overall results of design and development verification.
Design and Development Validation
Design and development validation has quite a few similarities to verification. Like verification, validation plans, methods, and acceptance criteria are to be defined before conducting. Like verification, applying statistical techniques to determine sample size may also be in order.
Design and development validation differs slightly based on the perspective and purpose. Validation means demonstrating that you have designed the correct product. Validation ensures that the medical device meets the customer needs and requirements.
Design and development validation must also be conducted with the product that is equivalent to the one in production. This means that products used for validation activities should be manufactured in the same manner with the same methods and techniques as what you anticipate for full production.
Upon completion of validation activities, results should be documented and demonstrate that the medical device meets customer requirements. This is another benefit of a traceability matrix to show the connections and relationships of all design and development elements. Design and development validations should also be used as a means to demonstrate effectiveness of risk controls and captured as part of your product risk assessment.
A design and development review is a steadfast way to check your work in this part of the process. It will provide overall results of design and development validation to show you whether you have successfully demonstrated and addressed the customer needs and requirements for your medical device.
Design and Development Transfer
Design and development transfer is not a single moment in time event. The entire purpose of design and development is to progress a medical device through product development so that it can eventually be ready for manufacturing. And throughout design and development, as noted above, there are certain activities at many of the stages that pertain to design and development transfer.
With that being said, there will be a moment in time when all of the product development is deemed complete. You have received the necessary regulatory permissions to go to market, and you are ready to complete transition to manufacturing. At such time, conduct a final design and development review of the entire product development project.
Control of Design and Development Changes
Design and development changes will occur throughout the entire product realization process. Any time a change occurs, you need to assess its impact on function, performance, usability, and safety. I generically describe this as “form, fit, function.”
Before design and development changes can be implemented, you must review, verify, validate (if required), and approve. While in design and development, one mechanism I suggest for documenting design and development changes is via a review. Post design and development transfer, design and development changes should be part of your document management / change management practices.
Design and Development Files
All of your design and development activities shall be documented and maintained in a design and development file. You may refer to this as a design history file or “DHF” (based on the FDA term for this).
My advice is to keep your design and development file living throughout the entire product realization process. I recommend using your design and development file up to date to represent the current product, including any and all changes.
Unfortunately, most medical device companies do not have systems in place to maintain living files for their design and development activities. What’s just as unfortunate is that the most common practice in the industry is for design and development files to be archived upon design and development transfer.
But realize that once transferred, there is a very good chance your medical device will have changes. Many times, these changes have an impact on design and development and should be assessed for form, fit, function, verification, validation, and risk.

ISO 13485 Purchasing
According to ISO 13485, your company should establish purchasing processes to make sure that the materials, components, and other products and services that you purchase meet defined specifications. This includes defining criteria for supplier evaluation, selection, and monitoring.
Supplier criteria should be risk-based and include:
- Supplier’s ability to meet requirements.
- Ongoing supplier performance.
- Impact the purchased goods have on your overall product quality.
- Risk purchased goods have on product risks.
- Criticality purchased goods are to the overall medical device.
For items being purchased, you should define specifications and requirements for acceptance. You should have documented agreements in place with your suppliers. The supplier agreement should address roles and responsibilities, identify supplier’s need to notify you of any changes to purchased goods, and applicable responsibilities to address QMS requirements.
Remember at the end of the day, you hold ultimate responsibility for ensuring your product quality and safety and for demonstrating compliance to QMS.
When you receive purchased goods, you need to verify these items meet your defined specifications. The type and level of verification should be risk-based; factors include supplier performance and criticality of the component. In the event purchased goods do not meet specifications, then you should document this via product non-conformance and attribute this to the supplier.
Purchasing and effective supplier management requires some level of traceability. A best practice for doing so is via an approved supplier list (ASL). The ASL should identify the specific goods that the supplier has been qualified to provide your company, supplier criticality, and status of supplier. You should document supplier monitoring activities and maintain supplier files. These activities should be logged on your ASL, as well.
A big question that comes up regarding suppliers is: Should you conduct on-site audits of suppliers?
Your supplier evaluation, selection, and monitoring activities should be commensurate with the risk a supplier poses to your products and business. The more critical the supplier to your overall product quality, the more importance should be placed on monitoring.
Yes, for some suppliers, frequent on-site audits may be a really good idea. For example, if you have outsourced production to a contract manufacturer, I would definitely advise that you audit this supplier before completing design and development transfer.
If at anytime in your relationship with a supplier there is a systemic issue that surfaces, this is a good use case for issuing a supplier corrective action request (SCAR).
![ISO 13485 production and service provision]() ISO 13485 Production & Service Provision
ISO 13485 Production & Service Provision
The purpose of production and service provision is to ensure that the manufacturing of your medical device is planned, executed, monitored, and controlled.
Essentially, compliance with ISO 13485 is about making sure your product meets defined specifications and that you have the necessary processes and environment for this to happen.
Control of Production & Service Provision
Production controls shall be risk-based. Your product controls shall include drawings, specifications, manufacturing instructions, labeling, packaging, and inspection procedures. The information you defined as part of design and development outputs and as part of your medical device file are essential for product controls.
For each product, you should define product release criteria, including any applicable activities for product delivery and post-delivery. Production controls should also indicate your infrastructure and work environment are appropriately documented and established for manufacturing of your products. This includes any production monitoring and measuring equipment that might be used.
You should have a record of product manufacturing. This record will vary depending on the risk and type of device. For some devices, the record may be an individual, serialized record. For others, the record may be lot-based or batch. In all cases, the production record shall identify quantity of product released for distribution, traceability to the medical device file, and evidence of verification and approval.
A note about software as a medical device (SaMD). Yes, it’s true that software is not manufactured in a traditional sense. However, these provisions also do absolutely apply to SaMD. Yes, even if your product is 100% software, there will still be a “manufacturer” of the product.
It’s common in the medical device industry to outsource manufacturing to contract manufacturing organizations. While the CMO may address many of the production and service provisions defined in ISO 13485, it is your responsibility as the medical device company whose name will be on the product to ensure this.
Make sure you have an agreement in place with your CMO that defines quality criteria, roles, and responsibilities. Refer back to the details described in the purchasing section for additional supplier management information.
Cleanliness of Product
If your product requires a certain level of cleanliness, this should be defined in a specification. And cleaning processes must be verified and/or validated and monitored.
Cleanliness of product applies if your medical device:
- Requires cleaning before use.
- Requires cleaning prior to sterilization.
- Process agents (e.g. machine oil) are to be removed during manufacturing.
Installation & Servicing Activities
If your medical device requires installation at point of use, then you must define installation requirements and specifications; these items will be part of the medical device file.
This includes establishing installation acceptance criteria and provisions to verify correct installation. These should have been captured when defining customer needs and during design and development. Records of installation shall be maintained as part of the product’s records.
Note: installation does not apply to all medical devices.
If your medical device requires servicing, then you must define servicing requirements, specifications, and procedures for doing so; these items are part of the medical device file.
Servicing also requires special attention to make sure the device meets its specifications after the product has been serviced. Servicing records shall be documented and maintained as part of the product’s records.
It is somewhat common for a medical device company to outsource installation and servicing to suppliers. As with other suppliers, installation and service suppliers shall be properly qualified, evaluated, monitored, and listed on your ASL.
Sterilization
For products that are sterilized and/or have sterile barriers, these processes must be validated prior to implementation. Refer to ISO 11607-1 and 11607-2 for specific sterilization requirements.
Sterilization criteria should be defined during design and development process. Sterilization process details and results of sterilization validation shall be part of the design and development file and medical device file.
When a production lot or batch is sterilized, records of sterilization shall be documented, traceable to the specific lot/batch, and part of the product’s manufacturing records.
Validation of Processes for Production & Service Provision
If the output of your manufacturing process cannot be verified, then this process must be properly validated prior to production. The purpose of manufacturing process validation is to demonstrate that the process achieves consistent results and that the product meets defined specifications and acceptance criteria.
Process validation shall include qualification of equipment used in the manufacturing process. Has the equipment been installed properly (IQ)? Does the equipment operate correctly and does the process produce acceptable results (OQ)? Does the process demonstrate long-term stability (PQ)?
There are also instances where process validation may apply, even if you can verify the output of your manufacturing process. There may be cases where you opt for efficiency and other business reasons where process validation would be beneficial.
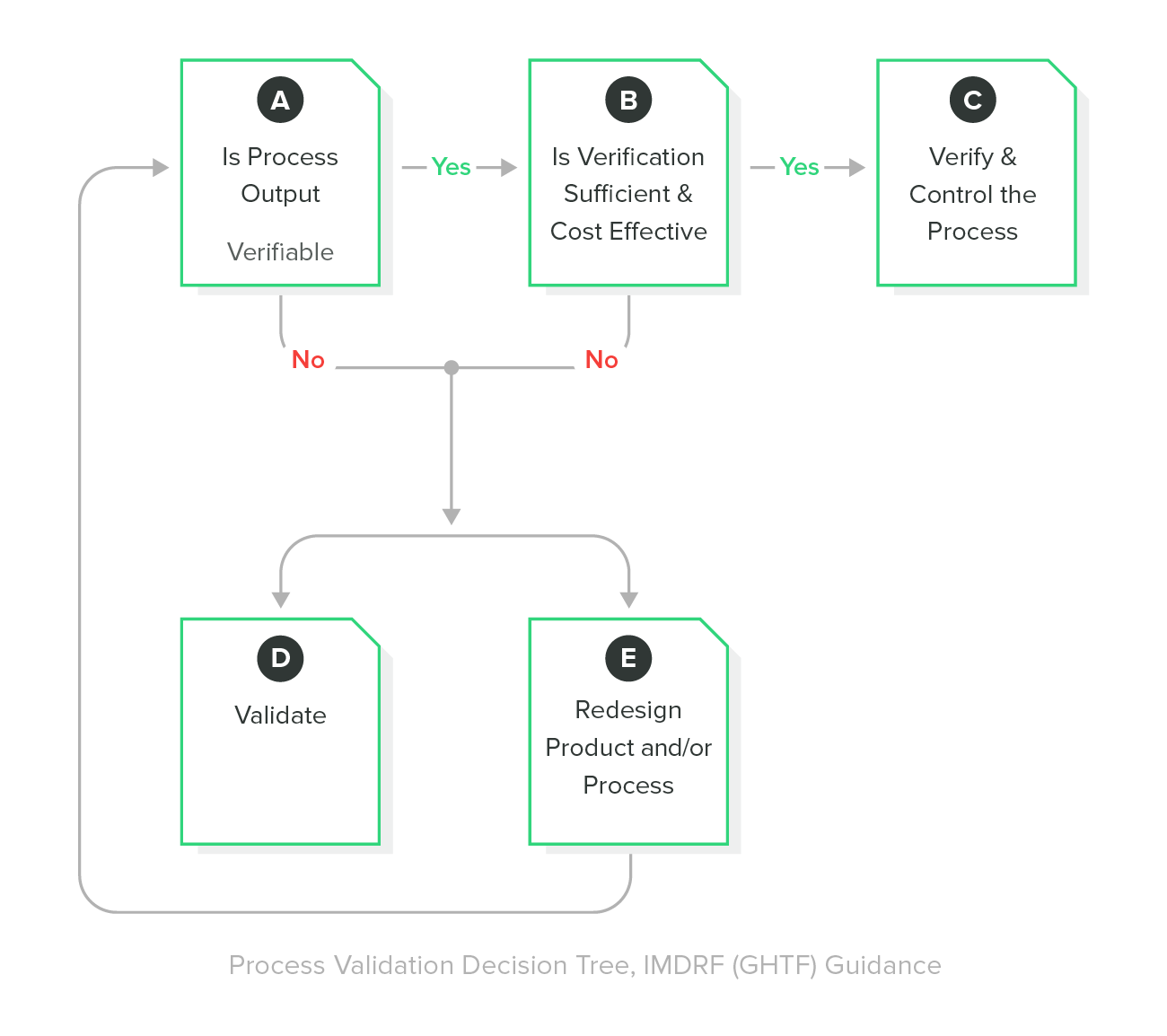
Process validation also should ensure training and qualification of personnel. Process validation methods, procedures, and acceptance criteria shall be documented, reviewed, and approved. Be prepared to use statistical techniques for sample size determination. There is an IMDRF / GHTF guidance for process validation that is a good resource to review.
Validation also applies when you use software in production, installation, and servicing activities. This software should be validated prior to initial use. The depth and detail for software validation under ISO 13485:2016 needs to be risk-based. Be sure to document the rationale and validation approach.
I recommend maintaining a list of all validated processes, often times referred to as a “Master Validation Plan” or “MVP.” In addition, you should define criteria for when re-validation is required, including if there is a process change. This should also be documented as part of document management/change control procedures.
Identification
ISO 13485 requires you need to be able to identify materials, components, sub-assemblies, and products throughout all stages of the product realization process and their disposition. This includes inventory, work in process, finished goods, distribution, installation, and servicing.
The purpose of identification is to know what products were manufactured and when.
Identification records shall be maintained for the product. One benefit of identification (although I hope this does not surface for you) is ability to track down products impacted by potential adverse events, recalls, and other quality events.
For finished medical devices, you may choose to identify with a unique serial number, lot number, or batch.
- Serial numbers usually apply to single product unit and is unique to that unit.
- As an example, a pump device would be assigned an individual serial number and be traceable to that specific unit.
- Lot numbers or batches typically apply to a certain number of products assigned to a group of products for which are common with specific properties.
- As an example, a lot number would be assigned to a group of needles manufactured against a particular order; all needles on that order would be assigned the same lot number.
Take special care to make sure that you can clearly identify and segregate any product returns from other products. You also need to make sure that any product found to be non-conforming is appropriately tagged and segregated from all conforming products.
For many markets today, medical devices require a unique device identification (UDI). Note, UDI criteria is often defined and included as part of the product’s labeling. I recommend referring to the IMDRF guidance on UDI for additional information, as well as the regulatory requirements for the particular markets.
Traceability
Traceability is similar to identification but applicable only after release from manufacturing. Not all medical devices require traceability. However, there are some products that definitely do, such as implantable medical devices. This varies depending on the specific regulations defined for products.
Note: This section on traceability is specific to the actual medical devices deployed for use and is different from design and development traceability matrix described earlier.
When traceability is required, records must be maintained when products are distributed. These traceability records must include name and address of when and where the product was shipped. Traceability helps you know very specific information about where your product is in the event of any pertinent quality events.
Customer Property
In the event you have customer property within your control, you need to have established processes on how to identify, verify, safeguard, and protect this property.
You should maintain records of these events. While not widely applicable, an example might be if a customer provides some type of measuring equipment to you to test.
Preservation of Product
You need to preserve and protect medical devices during processing, storage, handling, and distribution.
Define preservation specifications in documented processes and procedures. Examples might include packaging and shipping containers and specifying environmental criteria such as temperature and humidity.
![ISO 13485 monitoring and measuring equipment]() ISO 13485 Control of Monitoring & Measuring Equipment
ISO 13485 Control of Monitoring & Measuring Equipment
Throughout product realization, you are likely to use a variety of equipment to measure and monitor various aspects of your medical device. Any such equipment must be calibrated or verified to proven standards and criteria so that you know with confidence that the monitoring and measuring data is accurate and precise according to ISO 13485.
Such equipment shall also be labeled with identification, calibration status, and due date. You should maintain a record of all monitoring and measuring equipment, including calibration details. A good standard to consider for monitoring and measuring equipment is ISO 10012.
In some cases monitoring and measuring equipment may require process validation. This is especially true when software is used as part of monitoring and measuring. Refer to the earlier section on process validation.
ISO 13845 Measurement, Analysis & Improvement
How can you improve what you do not measure? In fact, I believe it was Peter Drucker who is credited with the quote,
"You can’t manage what you can’t measure."
If you refer back to the Plan-Do-Study-Act Deming Cycle shared towards the beginning of this guide, measurement is part of the ‘Do’ stage, analysis is part of the ‘Study’ stage, and improvement is part of the ‘Act’ stage.
During product realization, you defined product specifications. And throughout product realization, it is crucial to measure against specifications, analyze if there are trends, and make improvements. The same methodology also applies to the QMS as a whole.
The next few sections of this guide will focus on means of measurement, analysis, and improvement for your QMS and products.
Customer Feedback and Complaint HandlingI like to discuss customer feedback and complaint handling in ISO 13485 together because both are a means of receiving feedback about your products and services.
However, there is one important distinction, in my opinion—feedback is about being proactive, whereas a complaint is typically reactive in nature.
Do you recall the earlier discussion about customer needs and requirements? Feedback is about soliciting information to confirm that customer requirements have been addressed. Define procedures for gathering this feedback. And I encourage you to do so for all customer types for your products. Realize that the feedback you get may be positive, negative, or neutral.
The information you gather will be helpful too with post-production risk management.
Remember, risk management is a total product lifecycle process. And the information you learn post-launch will be helpful in ensuring your risk assessment, evaluation, and controls were properly captured.
According to ISO 13485:2016, by definition,
[A complaint is] written, electronic or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, usability, safety or performance of a medical device that has been released from the organization’s control or related to a service that affects the performance of such medical devices.
This definition is pretty broad and open to some interpretation. When you identify a complaint, there are certain minimum requirements that should be documented. You need to have a complaint handling procedure to define these requirements and process.
Complaint records need to capture:
- Product information, including product name, lot number, UDI, and any other identification information.
- Details of the issue, including date of the event.
- Date complaint received.
- Date and results of investigation.
- Any corrections and/or corrective actions taken.
- Correspondence with the person making the complaint.
Complaints generally have higher focus and visibility with regulatory agencies, especially during audits and inspections. With complaints, you need to assess whether or not further corrective action is required. As with feedback, complaints need to be part of your product risk management process.
Note that when you solicit proactive customer feedback, you may uncover an issue that needs to be logged as a complaint. If so, be sure to follow the complaint procedure for doing so.
Also realize that with both feedback and complaints, there are some events that might require further investigation and notification of regulatory authorities.
Generally speaking, these adverse events contributed to, or could have contributed to, serious injury or death. Hopefully you never encounter these types of situations. Regardless, you need to have established procedures that address how and what to do, if you receive adverse events.
Yes, I want you to take feedback and complaint processes seriously and realize how beneficial these post-market activities can be in analysis and improvement of your medical devices.
Please, please also take adverse event reporting very seriously.
Number one reason: this means there was some type of situation where a patient or end-user got injured. Secondly: you have a limited amount of time to react and respond to these events with regulatory agencies. So become very familiar with the specific regulatory requirements for the markets you are in with your products.
Internal Audit
Internal audits of your medical device quality management system should be considered prime opportunities for you to monitor the effectiveness of your processes. Again, referring to the Deming Cycle, internal audits are a means to do, study, and act.
Internal audits should be conducted at planned intervals and documented in some type of internal audit schedule. My advice is to spread your internal audits throughout the calendar year and to group similar/related processes.
A big mistake that I’ve seen way too many times is that companies conduct internal audits simply to check a box to satisfy the requirement. And oftentimes they will save all internal audits until the end of the year and rush to get through them. If this is your approach, you are not getting value out of your internal audits.
Document the results of your internal audits. And if you identify a systemic issue to be addressed, consider escalation in the form of a corrective or preventive action (CAPA) investigation.

Internal audits are an area to consider using a risk-based approach too. For example, if you determine certain processes are well-established, stable, and understood (corroborated with objective evidence, of course), this may be a lower risk area that requires less frequent audit cycles. On the other hand, the opposite scenario might mean a higher risk area where more frequent auditing would be warranted.
Control of Nonconforming Product
As discussed earlier in this guide, a critical part of product realization is to define specifications for your products. During manufacturing, if products do not meet these specifications, this is considered nonconforming product.
When nonconforming product is identified, it must be assessed and investigated. Disposition of a non-conformance should be risk-based in nature. My caution is to avoid the “use as is” disposition as much as possible. Why? If you allow “use as is,” does this mean your specifications are wrong? Maybe. And if so, consider escalating a corrective or preventive action investigation to explore the ramifications of changing the specification.
Rework is also a tricky disposition of a non-conformance. Tricky because if you do product rework, the rework instructions, processes, inspection criteria, etc., must be established. And the reworked product must meet the defined product specifications.
If you notice potential systemic nonconforming product issues, consider escalating a corrective or preventive action investigation.
Analysis of Data
Within measurement, analysis, and improvement, I have discussed customer feedback and complaints, internal audits, and nonconforming product. These are all processes where you should be tracking, trending, and analyzing data and KPIs. You should also be monitoring supplier performance as part of your analysis of data.
Often times when managing a QMS, and the corresponding data and information generated, we tend to be somewhat insulated and reactive in our data analysis. While it is good to definitely make sure that you are analyzing the effectiveness of your internal QMS and internal products, there is also value is analyzing other industry data. For example, consider analyzing other products in the industry similar to yours.
Corrective Action & Preventive Action
Yes, there should be a distinctive difference between corrective action and preventive action. And that major distinction is whether you are being reactive or proactive.
Yes, chances are you still lump both corrective action and preventive action into a single procedure and process that you refer to as “CAPA.” Is there any issue with this?
Well some would say yes--that corrective action and preventive action are different enough that these should be handled differently. Philosophically, I agree.
Practically and procedurally speaking, the process for conducting a corrective action investigation and preventive action investigation are for all intents and purposes very, very similar. ISO 13485 does differentiate between the two, and let me highlight the subtleties.
Corrective Action: eliminate the cause of nonconformities in order to prevent recurrence.
Preventive Action: eliminate the causes of potential nonconformities in order to prevent their occurrence.
Corrective Action “...shall be taken without any undue delay…”
Again, the major difference is that with corrective actions, the preceding systemic events have already occurred. With preventive actions, you have identified a potential systemic issue and gotten ahead of it beforehand with proactive measures.
Let me refer you to the Ultimate Guide to Corrective and Preventive Action (CAPA) for Medical Devices for a detailed step-by-step explanation.
As discussed throughout this guide, your medical device quality management system includes numerous processes and procedures. Procedures for customer feedback, complaints, internal audits, non-conformances, and supplier management are all means for you to measure, analyze, and improve these processes. And when the issue becomes a bigger, more systemic issue in nature, consider a corrective or preventive action.
How Greenlight Guru Streamlines ISO 13485 Certification
Greenlight Guru is the only ISO 13485-aligned QMS software designed by medical device professionals specifically and exclusively for the medical device industry. This means that by implementing Greenlight Guru, you’ll be addressing and complying with many of the ISO 13485:2016 requirements, automatically through the built-in workflows of the QMS.
Why is this important?
As I mentioned in the opening of this guide, unfortunately most in the medical device industry view QMS as a must-have to check boxes for compliance sake. Few understand and implement a QMS as a strategic business advantage.
Greenlight Guru views a QMS as the pinnacle solution for helping you run a better, more efficient medical device company. A QMS allows you to focus on the true quality of your products. And most importantly, a QMS should help your business always do what is best for the patients who will be recipients of your medical devices.
This is why the mission at Greenlight Guru is: To improve the quality of life.
The regulations are constantly changing in the medical device industry. Keeping up with the latest regulatory requirements can be daunting and a full-time job all by itself. This is why we are here.
Greenlight Guru focuses on keeping up on the latest updates with ISO 13485, FDA, ISO 14971, and so on, so that you can focus on improving the quality of your products and processes, knowing that the Greenlight Guru quality management system software has your compliance needs covered.
The Ultimate Guide to ISO 13485:2016 Quality Management System for Medical Devices


.png?width=250&height=324&name=UG%20ISO%2013485%20(1).png)



