QMSR is here! Is your quality system ready?

Solutions built for medical device teams to move faster, stay compliant, and grow with confidence.
Manage quality events, training, and documentation in one place.
Control documents, training, and change with traceability.
Track CAPAs, audits, and NCs with audit-ready records.
Qualify suppliers and manage parts with visibility.
Connect design, risk, and AI-powered traceability as you build.
Link needs, requirements, and verification to design controls.
Automate traceability from dev tools for faster releases.
Assess ISO 14971 risk as requirements and tests evolve.
Oversee studies with connected data, documents, and workflows.
Capture compliant clinical data with a validated medical EDC.
Plan and manage PMCF aligned to EU MDR expectations.
Collect clinical outcome data in-person or remotely on any device.
Built with regulatory insight to help medtech teams move faster with confidence.
Find issues faster and make smarter decisions with less manual work.
Tools and resources that help teams implement quickly and meet requirements.


A device master record (DMR) contains all of the information and specifications needed to produce a medical device from start to finish, including instructions for all manufacturing processes, drawings, documented specifications and labeling and packaging requirements.
The FDA regulations for DMR appear in 21 CFR Part 820.181, under subpart M which deals with record-keeping requirements for medical device companies. The device master record contains all of the information needed to produce the medical device from beginning to end.
While the device history record makes reference to specific lots, units, or batches of product, the DMR contains specifications for producing an individual device. You can think of it as the comprehensive instruction manual for building your device, starting with the design and ending with distribution.
FREE DOWNLOAD: Click here to download our Guide to DHF vs. DMR vs. DHR.
When the FDA audits your medical device company, you will be expected to produce a DMR that complies with 21 CFR Part 820.181. Here's the full text of that part for reference:
Each manufacturer shall maintain device master records (DMR's). Each manufacturer shall ensure that each DMR is prepared and approved in accordance with 820.40. The DMR for each type of device shall include, or refer to the location of, the following information:
(a) Device specifications including appropriate drawings, composition, formulation, component specifications, and software specifications;
b) Production process specifications including the appropriate equipment specifications, production methods, production procedures, and production environment specifications;
(c) Quality assurance procedures and specifications including acceptance criteria and the quality assurance equipment to be used;
(d) Packaging and labeling specifications, including methods and processes used; and
(e) Installation, maintenance, and servicing procedures and methods.
The main purpose of the device master record is to centralize a record of the production process in a way that distinguishes it from the design process.
A product development engineer might design a silicone part for a medical device, draw the design, and include it in the design outputs portion of the design history file, but the DMR would also include specific instructions for manufacturing the part, including what mold to use when casting the silicone, equipment specifications and production methods, and anything else required to produce the part according to the design.
It's so important to understand how the various record-keeping mandates set out by the FDA fit together, which you can learn more about in our eBook, Design History File (DHF) vs. Device Master Record (DMR) vs. Device History Record (DHR): What's the Difference? Here's a brief summary:
The FDA requires that your device master record is created in compliance with 21 CFR Part 820.40, which mandates document control throughout the QMS. As your manufacturing process evolves, your DMR will also change, and you'll need a review and approval process that validates any changes to the DMR.
If you are choosing to include a BOM as part of your DMR, you’ll need to ensure your BOMs are current, connected to key documentation and design controls. Like the BOM, DHF, and DHR documents, your DMR can either include or reference the required bill of materials.
That's where Greenlight Guru comes in — our QMS software includes built-in document controls that support electronic signatures, create and manage your items, export structured multi-level bills of materials, and allow your quality manager to review and approve changes to the BOM with the click of a button. You can easily link drawings and specifications in Documents to your BOM, export your BOM, and start putting together what you need for your DMR. 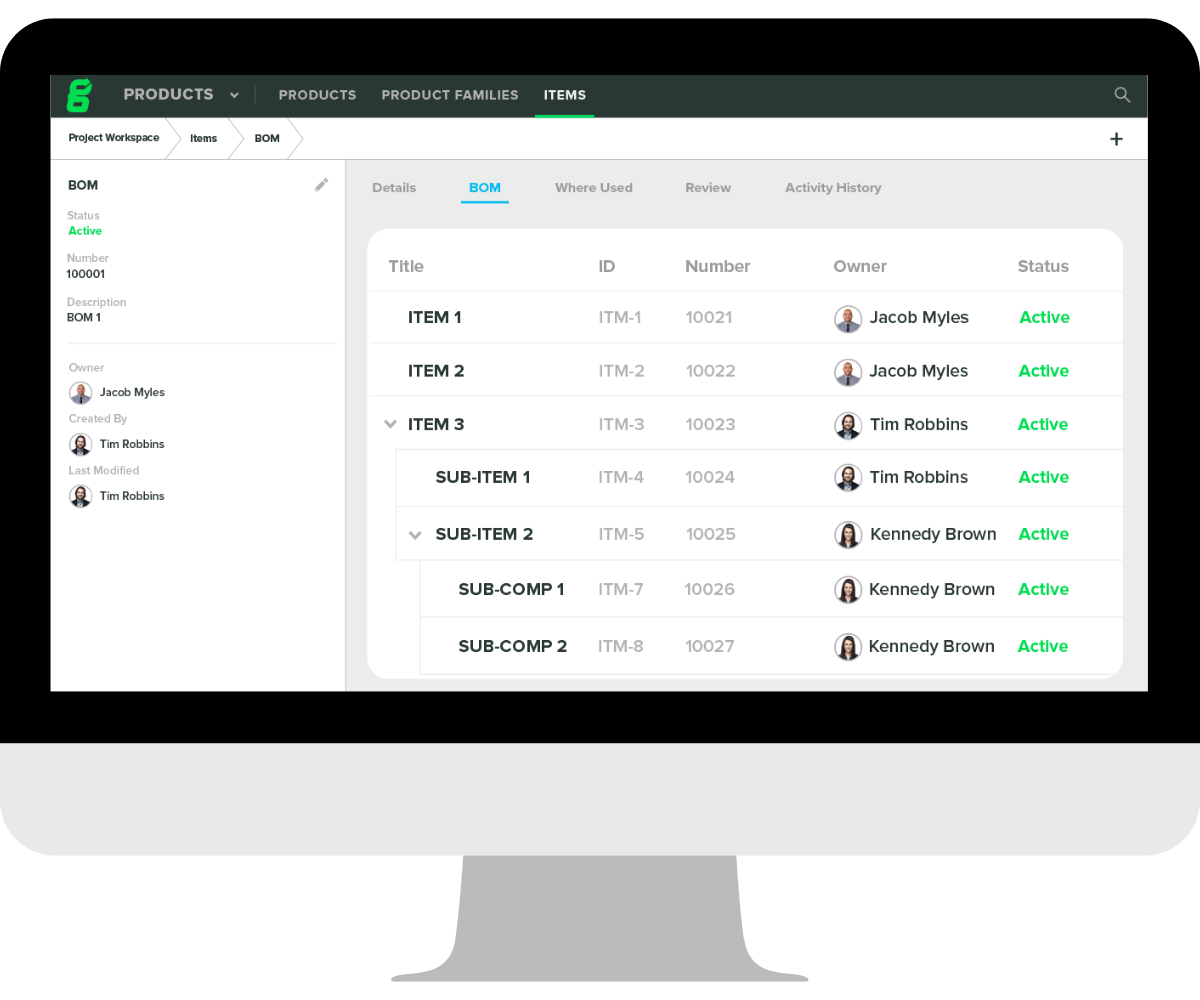
Instead of compiling a massive binder of documents, our QMS software allows you to securely store your medical device records where they can be easily referenced. You'll stay organized, save time and money, and make your next audit a breeze by using Greenlight Guru’s connected QMS solution.
Ready to get started? Contact us today for your free personalized demo of Greenlight Guru →
Looking for an all-in-one QMS solution to advance the success of your in-market devices and integrates your quality processes with product development efforts? Take an interactive tour of Greenlight Guru's Medical Device QMS software →




