








What Is This CAPA Guide About?
 Time and time again, the one quality management system process that nearly always presents the most challenges to medical device companies, regardless of shape and size, is CAPA. That’s right, Corrective and Preventive Action.
Time and time again, the one quality management system process that nearly always presents the most challenges to medical device companies, regardless of shape and size, is CAPA. That’s right, Corrective and Preventive Action.
And I suspect that your company might also be facing CAPA related challenges. Challenges relating to your CAPA process. Challenges related to your approach to initiating CAPAs. Challenges related to your CAPA closures. Challenges related to your the effectiveness of your CAPAs.
Yes, there are numerous challenges related to CAPA. This guide will explore those challenges.
More importantly, this guide will provide you with best practices and ways to improve your CAPA process and approach so that you can ultimately ensure the medical devices you design, develop, manufacture, and sell will be as safe and effective as possible for those patients that depend upon them.
In this ultimate guide to CAPA, I will share the following with you:
1. Regulatory Focus on CAPA and Related Processes
4. CAPA Process - Step by Step [Infographic]
5. How CAPA Connects to Other QMS Processes
6. Impact of Risk Management on CAPA
7. Shifting to Preventive Action
The main message that I want you to take home from reading this guide is this:
A robust and modern approach to CAPA is about shifting from reacting to situations and events to being proactive to address potential areas of concern before they become reality.
”It is clear from a review of FDA inspection findings that an organization’s CAPA process is the key to the maintenance and improvement of compliance with all regulatory QMS requirements,” said Ed Kimmelman, QMS technical consultant to AAMI and convener of the ISO/TC210, WG1, the developer of ISO 13485:2016."
FREE RESOURCE: Download our Ultimate Guide to Corrective and Preventive Action (CAPA).
Regulatory Focus on CAPA and Related Processes
You are probably well aware of the fact that FDA inspects medical device companies each and every year.
You might have even been a part of an FDA inspection. I also can imagine that you have also been through ISO audits. And if you have not been through any of these activities yet, I’m sure these will be part of your life at some point in the medical device industry. FDA inspections and ISO audits are a significant part of the medical device business.
Did you know that during a FDA inspection that your CAPA process will ALWAYS be evaluated?
You should also realize that CAPA process scrutiny is not just a FDA inspection thing. Your CAPA process will also always be evaluated during ISO audits.
Year after year, FDA reports data from inspections. And year after year, companies receive FDA 483 inspectional observations for “lack of or inadequate procedures” for CAPA.
In fact, every year going back to 2010, the #1 reason companies receive 483s is because of CAPA.
“Procedures for corrective and preventive action have not been [adequately] established.”
It should also be noted that you will notice a similar trend with respect to FDA issued warning letters. Yes, CAPA is often the #1 quality system citation in warning letters. Below is a table from 2016 FDA quality system data analysis.
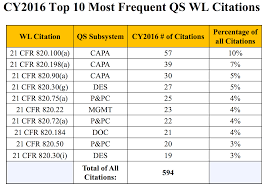
(Are you interested in viewing FDA inspectional data yourself? Here is a link to check out FDA inspectional data and another to review FDA quality system data from inspections and warning letters.)
Unfortunately, there is not similar data available ISO 13485:2016 audits. But based on my ISO audit experiences and the hundreds of medical device professionals I have spoken to about this, CAPA is an equally big deal during ISO audits.
Good news. There are guidances available to help you.
FDA has published “Guide To Inspections of Quality Systems” (often referred to as “QSIT”) where they tell you how they will conduct an inspection. For the inspection guide on CAPA, refer to this link.
There is also a published guidance available from IMDRF regarding corrective action and preventive action and related QMS processes.
This regulatory emphasis on CAPA all makes sense. Some would argue that CAPA may be the single most important process within your QMS.
Regardless of the ranking, it is critical to realize how many of your QMS processes “feed” into the CAPA process and how the results of CAPAs impact the health of your overall QMS. (I’ll share more about how and which QMS processes connect with CAPA later on in this guide.)
Note: If you have a medical device on the market at your company, Greenlight Guru’s postmarket workflows allow you to streamline your quality processes like CAPAs, Nonconformances, Complaints, Audits and more in a single, connected software platform. Learn more about our CAPA Management software here.
What is CAPA?
Okay, it probably makes sense to take a bit of time to discuss what is CAPA before diving too deep into all the problems.
Let me start with sharing how FDA and ISO 13485 defines the CAPA meaning.
FDA requirements for CAPA are defined in 21 CFR Part 820.100 Corrective and Preventive Action:
(a) Each manufacturer shall establish and maintain procedures for implementing corrective and preventive action. The procedures shall include requirements for:
(1) Analyzing processes, work operations, concessions, quality audit reports, quality records, service records, complaints, returned product, and other sources of quality data to identify existing and potential causes of nonconforming product, or other quality problems. Appropriate statistical methodology shall be employed where necessary to detect recurring quality problems;
(2) Investigating the cause of nonconformities relating to product, processes, and the quality system;
(3) Identifying the action(s) needed to correct and prevent recurrence of nonconforming product and other quality problems;
(4) Verifying or validating the corrective and preventive action to ensure that such action is effective and does not adversely affect the finished device;
(5) Implementing and recording changes in methods and procedures needed to correct and prevent identified quality problems;
(6) Ensuring that information related to quality problems or nonconforming product is disseminated to those directly responsible for assuring the quality of such product or the prevention of such problems; and
(7) Submitting relevant information on identified quality problems, as well as corrective and preventive actions, for management review.
(b) All activities required under this section, and their results, shall be documented.
Further, in the FDA QSIT guide, they describe the purpose of CAPA as:
The purpose of the corrective and preventive action subsystem is to collect information, analyze information, identify and investigate product and quality problems, and take appropriate and effective corrective and/or preventive action to prevent their recurrence. Verifying or validating corrective and preventive actions, communicating corrective and preventive action activities to responsible people, providing relevant information for management review, and documenting these activities are essential in dealing effectively with product and quality problems, preventing their recurrence, and preventing or minimizing device failures. One of the most important quality system elements is the corrective and preventive action subsystem.
In ISO 13485:2016, CAPA is addressed in two separate clauses: 8.5.2 Corrective Action and 8.5.3 Preventive Action.
8.5.2 Corrective action
The organization shall take action to eliminate the cause of nonconformities in order to prevent recurrence. Any necessary corrective actions shall be taken without undue delay. Corrective actions shall be proportionate to the effects of the nonconformities encountered.
The organization shall document a procedure to define requirements for:
a) reviewing nonconformities (including complaints);
b) determining the causes of nonconformities;
c) evaluating the need for action to ensure that nonconformities do not recur;
d) planning and documenting action needed and implementing such action, including, as appropriate, updating documentation;
e) verifying that the corrective action does not adversely affect the ability to meet applicable regulatory requirements or the safety and performance of the medical device;
f) reviewing the effectiveness of corrective action taken.
Records of the results of any investigation and of action taken shall be maintained (see 4.2.5).
8.5.3 Preventive action
The organization shall determine action to eliminate the causes of potential nonconformities in order to prevent their occurrence. Preventive actions shall be proportionate to the effects of the potential problems.
The organization shall document a procedure to describe requirements for:
a) determining potential nonconformities and their causes;
b) evaluating the need for action to prevent occurrence of nonconformities;
c) planning and documenting action needed and implementing such action, including, as appropriate, updating documentation;
d) verifying that the action does not adversely affect the ability to meet applicable regulatory requirements or the safety and performance of the medical device;
e) reviewing the effectiveness of the preventive action taken, as appropriate.
Records of the results of any investigations and of action taken shall be maintained (see 4.2.5).
The Problems with CAPA Management
As shared so far, you should have a better understanding as to visibility of your CAPA process and why having a good approach is so important to the health and success of your medical device company.
Let me share a few reasons why there are problems with CAPA, including:
- Lack of cross-functionality
- Reactive instead of proactive
- Overuse versus underuse
- Poor root cause determination
- Poor definition of a CAPA process
Lack of cross-functionality
Generally speaking, CAPA management is most often a process that is owned by the quality function within a medical device company. Quality usually holds the keys and makes the determination as to when a CAPA is required.
This makes sense since the quality organization is responsible for your company’s QMS implementation and effectiveness.
If the quality function unilaterally makes the decisions as to what does and does not become a CAPA, does this make sense? Probably not.
Does the quality organization have visibility into all the sources of data and information to make these decisions? I doubt it.
Nearly every single medical device company I’ve ever worked with and know about is missing a “single source of truth” in order to make informed decisions about when and what to do. (I’ll share more about this later on in this guide.)
What should you do to address this concern?
Realize that a CAPA is seldom an issue that only impacts the quality function. A CAPA almost always is cross-functional in nature and involves many other groups and functions of your company.
A recommended best practice is to establish a cross-functional team to review and discuss issues that may be candidates for a CAPA. Such a team is often called a “management review board” or “MRB” and is comprised of representatives from quality, regulatory, operations, and engineering.
A MRB should meet frequently, probably at least weekly, and review issues related to customer feedback, complaints, audits, nonconformances, etc. A MRB then assesses each of the issues and makes determinations as to which should be elevated to a CAPA.
Further, I recommend that your MRB maintains a list of the open issues, establishes an agenda for their meetings, and documents minutes from the decisions made. The goal of the MRB is to review, assess, evaluate, prevent, correct, and control quality issues.
Once a CAPA is issued, you also need to realize a couple of key points. A CAPA is a project that will require resources to address. Once again, this is an area where cross-functionality is absolutely essential. All too often in medical device companies, CAPAs are not treated with equal status and importance as other initiatives, such as new product development.
This is one of the reasons how and why CAPAs linger and are ultimately ineffective.
Reactive instead of proactive CAPA management
The IMDRF has a guidance document on corrective action and preventive action and related QMS processes. Within this guidance, there is a profound statement about what CAPA is that I want to share with you.
The acronym “CAPA” will not be used in this [guidance] document because the concept of corrective action and preventive action has been incorrectly interpreted to assume that a preventive action is required for every corrective action.
This document will discuss the escalation process from different “reactive” sources which will be corrective in nature and other “proactive” sources which will be preventive in nature. The manufacturer is required to account for both types of data sources whether they are of a corrective or preventive nature.
I find this profound for one major reason.
This guidance was published in 2010, yet still today, medical device companies seem to place most of their CAPA efforts on correcting issues rather than preventing them in the first place.
Is this because of the overuse of the acronym “CAPA”?
Perhaps.
But I suspect that this is more about a mindset, or philosophy. We are more inclined to address known problems rather than investing resources to prevent problems from ever happening.
To quote one of Murphy’s Laws:
“There is never enough time to do it right the first time, but there is always enough time to do it over.”
It is very common for CAPAs to not be taken seriously in an organization. Most view CAPA management as part of the quality department and not applicable to them.
CAPAs often take a back seat to other projects and initiatives deemed more important. However, CAPAs are enterprise-wide and impact the entire organization. CAPAs are a primary indicator of the health of your company’s QMS.
But what if instead of waiting for things to happen, you could change your approach to be more proactive?
There is a great quote from hall of fame basketball coach John Wooden that applies here.
“If you don’t have time to do it right, when will you have time to do it over?”
Sadly, CAPAs are more times than not reactions to issues that have already occurred. And it’s only after the issues have happened do medical device companies seem to have the time to do it right.
This mentality and approach is wrong. This way of doing things causes a significant strain on medical device businesses. You seldom plan for issues to happen.
I get it. We are all doing the best that we can in order to get medical devices to market. And once we launch these products, we have established processes to gather customer feedback and complaints, identify nonconformances, and auditing. We use these downstream processes to help identify problems and then take necessary actions by issuing CAPAs.
As a result, this reactive approach is also somewhat understandable.
Why?
The medical device industry does not have easy access to data to shift this mindset and approach. The methods used to evaluate situations and issues is more times than not a lagging indicator, rather than a leading indicator.
We need to have systems in place to assess and identify issues before they become problems.
We need to dedicate resources to constantly make our medical devices safer and better.
CAPA Overuse vs CAPA underuse
Let me share another systemic problem regarding CAPA I have observed. Simply stated, a CAPA quality process is often at one extreme or another. Either CAPA is overused or it is underused.
Let me elaborate and explain the overuse and underuse scenarios.
Imagine you receive a complaint on one of your medical devices. Or maybe you have a nonconformance. Does this singular event warrant issuing a CAPA investigation?
It might.
However, I’ve seen where some companies issue CAPAs in nearly all situations like this. And before long, there are dozens and dozens of CAPAs, in addition to the complaints, nonconformances, and other quality events.
This practice is overuse of the CAPA process.
This practice creates an undue burden on a medical device company. Conventional wisdom might indicate that issuing a CAPA will lead to improving an issue. But in reality, overuse of CAPA actually results in more significant challenges.
When overused, there are too many CAPAs and strains on resources. CAPAs either linger for months and months or are rushed to complete, without properly addressing underlying issues.
The other extreme is underuse of CAPA process.
When this happens, companies do not use CAPA nearly enough. There may be quality events happening, yet these do not result in CAPAs.
Both overuse and underuse of CAPAs are problematic.
Is there a magic number of CAPAs that a company should have open? No, there is not. So many factors influence if and when a CAPA should be issued.
CAPA should be reserved as a process to deal with potential systemic issues.
Poor root cause determination
Root cause determination is one of the biggest areas of concern, especially as it relates to CAPA management.
From my observations, there is very little time spent actually determining a root cause. Rather, the prevailing practice is to more or less restate the problem statement / issue description and capture this as the root cause.
The problem statement and issue description is hardly ever the true root cause.
To correct or prevent a problem, you have to define the root cause. Otherwise, your efforts are likely to miss the mark. Yes, you may take care of the problem for the short term. But without getting to the root, the issue has a high potential to happen again. And again.
Defining a root cause takes some effort and time. This is not something you can rush and easily slap together.
You need to take the necessary steps to properly define root cause. Without doing so, any actions you define within your CAPA are likely to miss the mark.
To say this another way, if you do not drill down to properly define the root cause, the issue you are trying to address will likely still be present, despite the actions you take.
Good news. There are numerous root cause tools and techniques, if applied properly, will make a huge improvement to your CAPA process. But deploying root cause requires a bit of discipline and embracing cross-functionality.
I’m a big fan of the “5 whys” approach because it is relatively easy to understand and can be quickly implemented. Let me briefly illustrate this technique.
Issue: The device breaks during use.
Why? → The device is being exposed to extreme forces at point of use.
Why? → The use technique at facility ABC is different than intended.
Why? → Facility ABC did not receive in-service training.
Why? → There is no resource assigned to provide training for that facility.
Why? → No process to confirm that all use facilities require in-service training before products can be sold to a facility.
Note, with “5 whys”, the number of why questions is less important, as long as you are getting to an actionable root cause; may be 3 whys or could be 10 whys.
Root cause determination can take quite a bit of time and require cross-functional resources, if done correctly. This should not be rushed through in a couple of hours by a single person.
Regardless of the tools you choose, do make sure that you improve your root cause determination. Doing so will help ensure that your CAPAs are more effective in the long run.
Ignoring or choosing to continue with status quo on root cause will mean CAPAs for similar issues will crop up over and over and that your efforts will not be effective.
Poor definition of a CAPA process
CAPA process definition is a huge issue for medical device companies. If you recall, this issue is the single biggest reason FDA issues 483 observations to medical device companies year after year since 2010. (Refer to the Regulatory Focus section in this guide for more about FDA and ISO.)
This usually means one of three things:
- Your defined CAPA process is not being followed.
- Your defined CAPA process is not compliant.
- You have not defined a CAPA process.
There might be another issue lurking too. And this would be that you have either not utilized your CAPA process appropriately or are overusing CAPA and not getting things done.
Bottom line: This is a huge problem. Companies do a very poor job when it comes to effectively managing CAPAs. And yes, the root cause of this usually lies with poor definition of your CAPA process.
Based on the historical data of FDA inspections and issued 483 observations, I would bet that your CAPA process and practices need work.
And because of this, I would like to offer a step by step description of a CAPA process that will be step in the right direction.
CAPA Process in 15 Steps
First and foremost, realize that your CAPA process is best utilized for systemic issues. Again, keep in mind all the items discussed throughout this guide. Make sure first and foremost that you are properly leveraging CAPA appropriately.
Assuming this is the case, I’d like to suggest some high-level steps to consider for your CAPA process.
- Create CAPA request and submit for review. Identify sources of CAPA worthy issue.
- Review CAPA request. Determine whether or not issue requires CAPA investigation.
- Accept or reject CAPA request.
- If accepted, issue and initiate CAPA.
- Finalize CAPA sources (i.e. products, processes).
- Determine CAPA cross-functional team.
- Identify any immediate actions and corrections required.
- Investigate and capture findings.
- Determine root cause(s).
- Specify corrective and/or preventive action plan.
- Complete action plan.
- Review CAPA for completeness.
- Approve CAPA actions.
- Specify steps for effectiveness verification.
- Verify effectiveness of CAPA.

How CAPA Connects to Other QMS Processes
As noted throughout this guide, CAPA is an important process for your medical device company.
In fact FDA states in their QSIT guide:
“One of the most important quality system elements is the corrective and preventive action subsystem.”
A solid CAPA process is foundational and an indicator of the health of a medical device company.
While a healthy CAPA process is key to a healthy QMS, it is equally important to understand how other QMS processes connect with CAPA. Some of the key QMS processes related to CAPA include:
- Complaint Handling
- Customer Feedback
- Nonconforming Product
- Production & Process Controls
- Supplier Management
- Audits
- Design Controls
- Management Review
Complaint Handling
Once a product is launched, one of the first areas that provides an indication for product opportunities and issues are your customer feedback and complaint handling processes.
From a FDA perspective, complaint handling is a very critical process. How and what you do when you receive complaints is important. Why? If a complaint caused an adverse event, then this could require a thorough investigation and reporting to FDA and other regulatory agencies.
Even if a complaint is not an adverse event in nature, it still is an indicator of your product’s performance. Investigating complaints requires a robust process. A robust complaint handling process is very similar in concept to a CAPA process, as described earlier.
Define the issue, determine root cause, establish an action plan to address and correct the situation.
Generally speaking, complaints are often times more reactive in nature. To say another way, a complaint is usually an event that has already happened. You seldom seek out complaints. Instead, you are contacted after a complaint has occurred. When you learn of the complaint, addressing the immediate issue is corrective in nature.
But here is where understanding the connection between complaint handling and CAPA processes is important. Just because you have a complaint does not mean you automatically need to issue a CAPA.
As noted, if your complaint process is robust, it will help ensure the complaint issue is properly addressed. Remember that CAPA is ideal for addressing systemic issues. Monitoring complaints and analyzing the underlying issues is important for determining if you have systemic product issues to address.
Let me illustrate with an example.
You get a call from a doctor that your device has an issue with a connector. You conduct a complaint investigation and address the situation for the doctor.
When you review other complaint records and analyze the data, you discover that there have been other connector issues with this product line, as well as a couple other product lines.
This analysis uncovers a potential systemic issue regarding connectors. This type of scenario is CAPA worthy.
And while addressing a singular complaint is somewhat reactive in nature, analyzing data for other similar issues is a way for you to be more proactive.
The approach, however, that most seem to take in this type of scenario is to wait for the connector issue to surface multiple times as individual complaints and then issue a CAPA. This approach is very reactive.
Customer Feedback
Customer feedback is a concept that was introduced to the medical device industry with the emergence of ISO 13485 several years ago. The basic premise of customer feedback is that you are seeking feedback on your products, rather than just waiting on the feedback to come to you.
Very few companies have successfully implemented a good customer feedback process. Most rely heavily on complaint handling as a primary means of getting feedback. And as noted above, complaints are reactive in nature.
Idealistically speaking, implementing a robust customer feedback process will help you to identify opportunities for improvement for your products before product issues surface. A thorough customer feedback process is about being proactive. It’s about adopting an approach of continuous improvement.
How does customer feedback relate to your CAPA meaning? Again, it’s about evaluating and analyzing data. It’s about understanding where there are opportunities for improvement. When you identify these opportunities, then evaluate if addressing them is CAPA worthy. Do you see how this approach is being proactive?
Nonconforming Product
When you launch your device into the market, you need to have processes in place to evaluate and ensure that the products conform to established specifications prior to release. In the event that your product does not meet the defined specifications, then this means the product is nonconforming.
A solid nonconformance process is a good proactive measure; potential product issues should be identified before the product is shipped. A solid nonconformance process identifies the issue, incorporates root cause determination, and includes action plans to address the situation.
Without a solid nonconformance process in place, you are likely setting your company up to receive more product complaints.
If an nonconformance issue happens one time, this is not necessarily CAPA worthy. Remember, the meaning of CAPA is a process to address systemic issues.
Like the complaint example cited above, analyzing nonconformance data is a proactive means to identify whether or not systemic issues are prevalent. If so, then yes, by all means, issue a CAPA to address the issue. Taking this type of approach is being proactive to address situations instead of just reacting to them after the fact.
Production & Process Controls
Every medical device requires production and process controls. Production and process controls include documented steps required to manufacture medical devices. This includes inspection procedures.
Production and process controls are provisions you establish to ensure that the device is manufactured according to established specifications--specifications established to ensure the product is safe.
Yes, production and process controls also apply to software-based medical devices. While I realize software as a medical device (SaMD) products are not “manufactured” per se, you do need to have established processes to ensure specifications are met and inspection criteria is defined. You must ensure that your SaMD is safe.
Technically speaking, product issues identified during the production process should be captured as nonconformances. A nonconformance is the initial means to address issues with product not meeting established specifications. And as noted above, if the nonconformance is systemic in nature, then issuing a CAPA investigation is a best practice.
There could also be issues with the production related processes. If you identify issues with production processes, then you need to determine a course of action. More times than not, you likely can tackle these via your change management process. However, if the issues are more systemic in nature, then consider a more thorough CAPA investigation.
Supplier Management
Supplier management involves qualifying, evaluating, and monitoring the performance of your suppliers.
If you identify issues with items purchased from suppliers, then the first step is to issue a nonconformance. Again, a nonconformance is a means to document an issue when an item does not meet established specifications. When this happens, you need to determine, within the nonconformance documentation, whether to use as is, rework, or return to supplier.
If this happens a time or two, then managing the issue via nonconformance is likely sufficient. If a supplier has repeat infractions of failing to provide items that meet your specifications, then this is systemic. Yes, you guessed it. A CAPA investigation would then be a best practice.
But what type of CAPA? Should this be handled internally by your company only? Maybe. Maybe you did a poor job of establishing the specifications.
There might also be times when you need to issue a SCAR (supplier corrective action request) to your supplier. Issuing a SCAR is elevating the seriousness of the issue both within your company and (hopefully) with your supplier.
If you issue a SCAR, you should track the actions related to this within your CAPA process, identifying the type as supplier related.
Audits
In the medical device industry, we are subjected to all kinds of audits and inspections: ISO audits, FDA inspections, supplier audits, internal audits, and so on.
Most of us dread the thought of being audited. Audits are time-consuming and strain resources. Audits uncover issues that, often times, need to be addressed--especially if discovered by a FDA inspector or ISO auditor.
From another point of view, audits can be great opportunities for making improvements. Audits can be great opportunities to discover potential issues before they become problems.
Let me talk a bit about internal audits specifically.
Internal audits comprise of activities to self-assess and self-evaluate your QMS effectiveness. However, too many companies do not take this approach with respect to internal audits. Many companies do internal audits because they have to--a check box activity.
But internal audits, done properly, should be more rigorous and challenging than any other ISO audit or FDA inspection. In fact, this should be your goal of your internal audit program.
As you conduct internal audits, you may uncover items to address. If these issues are systemic in nature, then these issues are likely candidates for a more thorough CAPA investigation.
Design Controls
Design controls are all about ensuring that you have demonstrated, proven, and documented that your medical device is safe, effective, and meets the needs of the end-user.
If you have a significant number of product nonconformances and/or product complaints, then I suggest you take action. To say it another way, volume of nonconformances and complaints are a direct reflection of your design controls process. Design controls best practices are the #1 way to reduce product-related quality events.
I’ve already discussed the need to issue CAPA investigations if you have systemic nonconformances and systemic complaints.
If this happens to be the case, I highly recommend that you take another look at your design controls practices as well. I highly recommend you consider issuing a CAPA.
Management Review
Management reviews are times when your executive leadership should be evaluating the effectiveness of your quality management system. Management reviews are times to assess the health of your medical device company.
Reviewing customer feedback and complaints, nonconforming products, audits, and supplier performance are all invaluable inputs to management review. And as noted above, CAPA is one of the most important quality system elements. As such, CAPA effectiveness is the keystone of management review.
CAPA and the QMS Summary
I’ve shared some insights about how QMS processes for customer feedback, complaints, nonconforming product, production and process controls, supplier management, audits, design controls, and management review interact and relate to CAPA.
If these other QMS processes are effective, then congratulations! Your quality system is likely right-sized and effective.
If not, then do something about it! Establish an effective CAPA process to address your systemic issues, and get to a state of QMS effectiveness.
.png)
![Impact_Risk_Management_CAPA_6]() Impact of Risk Management on CAPA
Impact of Risk Management on CAPA
 Impact of Risk Management on CAPA
Impact of Risk Management on CAPARisk management is a process that is very much here to stay in the medical device industry. On the product side of risk, ISO 14971 continues to be the cornerstone of identifying, assessing, evaluating, and controlling risks as a means to ensure medical devices are as safe and effective as possible.
How does CAPA play a role in product risk management?
Many CAPAs will impact medical device products in some way, shape, or form. Whether addressing a design issue, how the device is used, dealing with specifications, or manufacturing processes, CAPA actions need to consider and address risk management.
And it’s not enough to just check a box on a CAPA form. Addressing risk requires reviewing documented product risk management to determine if the issues within the CAPA are defined accurately. If not, then update your risk management accordingly.
From a product side of risk management, this interaction with CAPA is so important. Recall that ISO 14971 establishes risk management as a total product lifecycle process. However, many do not truly keep their risk management files up to date and current.
Another risk concept that was formally introduced to the medical device industry with the publication of ISO 13485:2016 is “risk-based QMS”. What does this mean and how does this relate to CAPA?
Yes, it’s true that ISO 13485:2016 does make reference to ISO 14971. And from the product side of things, that makes complete sense. The references also infer that a risk management process and framework is well-defined and well-established by ISO 14971 and that this framework is also applicable to your QMS.
Further, I shared earlier how CAPA is largely regarded as one of, if not the, most important quality system elements.
From my perspective, the concept of a risk-based CAPA process becomes foundational to the health and success of your medical device company. There are a lot of factors to consider with respect to apply risk concepts to CAPA. Let me provide a few suggestions.
Risk-based decision making is almost approaching cliche status these days. However, your CAPA process should incorporate the concept of risk-based decision making from the moment you learn of a quality event (such as a complaint or nonconformance).
Does the quality event require a formal CAPA investigation? This is an example of risk-based decision making. And I’ve discussed above about when you should consider a CAPA.
Once a CAPA request is submitted, then the decision as to whether or not to proceed with a CAPA should also be a risk-based decision.
After a request is accepted as a formal CAPA, then identifying the priority and urgency are also important and also should be risk-based decisions.
Ensuring all products, processes, and sources are identified within a CAPA are key risk elements. In other words, when you issue a CAPA, don’t be too myopic; consider if the issue to be addressed is also prevalent with other products and processes. Be holistic. (Taking this approach could actually reduce the volume of CAPAs and be a way for you to shift to being proactive, rather than reactive.)
Drilling down and identifying root cause is also a risk-based approach. If you do a poor job with root cause, then the issue has a likelihood of happening again.
Once root cause is determined, then the actions you take are key to controlling, reducing, and mitigating risks. The actions you take, however, are directly related to the identified root cause.
Upon completion of actions, you will need to verify the effectiveness of those actions. This verification step is very crucial because this should be when you determine and confirm, with objective evidence, that the CAPA has been addressed successfully.
To say it another way, your entire CAPA process should be entirely risk-based, from the moment a request is made until you have verified the effectiveness of the actions taken.
Shifting to Preventive Action
It’s now time to shift your approach with CAPA. It’s time to reduce your reliance on reacting to situations, events, and issues and to shift to being proactive.
Being reactive, or correcting issues, creates unnecessary challenges to your business and strains your invaluable resources. Being reactive is a sort of “victim” mindset. We should not be waiting for things to happen.
Instead you should be seeking ways to improve preventing issues from ever happening in the first place. You should be using your QMS as it was intended: to focus on “True Quality” of your QMS processes, and most importantly the medical devices that are intended to improve the quality of life!

Single Source of Truth
I know, making the shift from “corrective action” to “preventive action” is going to be hard.
There are numerous reasons as to why. Maybe the biggest factor to being proactive is related to all the sources of data and information.
How many different systems does your company have to address all the quality events?
I recently heard a story from a medical device professional that his company had about 10 different systems in place. One for customer feedback and complaints. Another for nonconformances. And so on.
None of these systems “talked” to one another. Each of these systems had different owners. The functional groups did not collaborate. No one had total visibility of all of these quality data sources.
I’m afraid that this story is not an isolated case. The overwhelming majority of medical device companies are in a similar situation.
Imagine a different existence. Imagine that you had one system where your entire QMS was managed. Imagine that you had one system where you could manage design controls, risk management, nonconformances, customer feedback, complaints, audits, and CAPAs.
And now imagine that this one system connected all the data related to your products and processes so you could for the first time truly understand how and what to do.
I know it sounds too good to be true.
Think about how great it would be to have a QMS that works. A QMS that is healthy. A QMS that is designed for how your business operates. A QMS that extends beyond just dealing with compliance and one that focuses on “True Quality”.
Know that you can have this one system. A single source of truth for your QMS and all the data, information, documentation, and records.
Know that you can have this one system, designed specifically for the medical device industry by expert medical device professionals.
Learn more about Greenlight Guru's CAPA Management Software by scheduling your demo today.
Looking for an all-in-one QMS solution to advance the success of your in-market devices and integrates your quality processes with product development efforts? Click here to take a quick tour of Greenlight Guru's Medical Device QMS software →
The Ultimate Guide To CAPA






