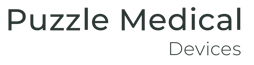QMSR is here! Is your quality system ready?

Solutions built for medical device teams to move faster, stay compliant, and grow with confidence.
Manage quality events, training, and documentation in one place.
Control documents, training, and change with traceability.
Track CAPAs, audits, and NCs with audit-ready records.
Qualify suppliers and manage parts with visibility.
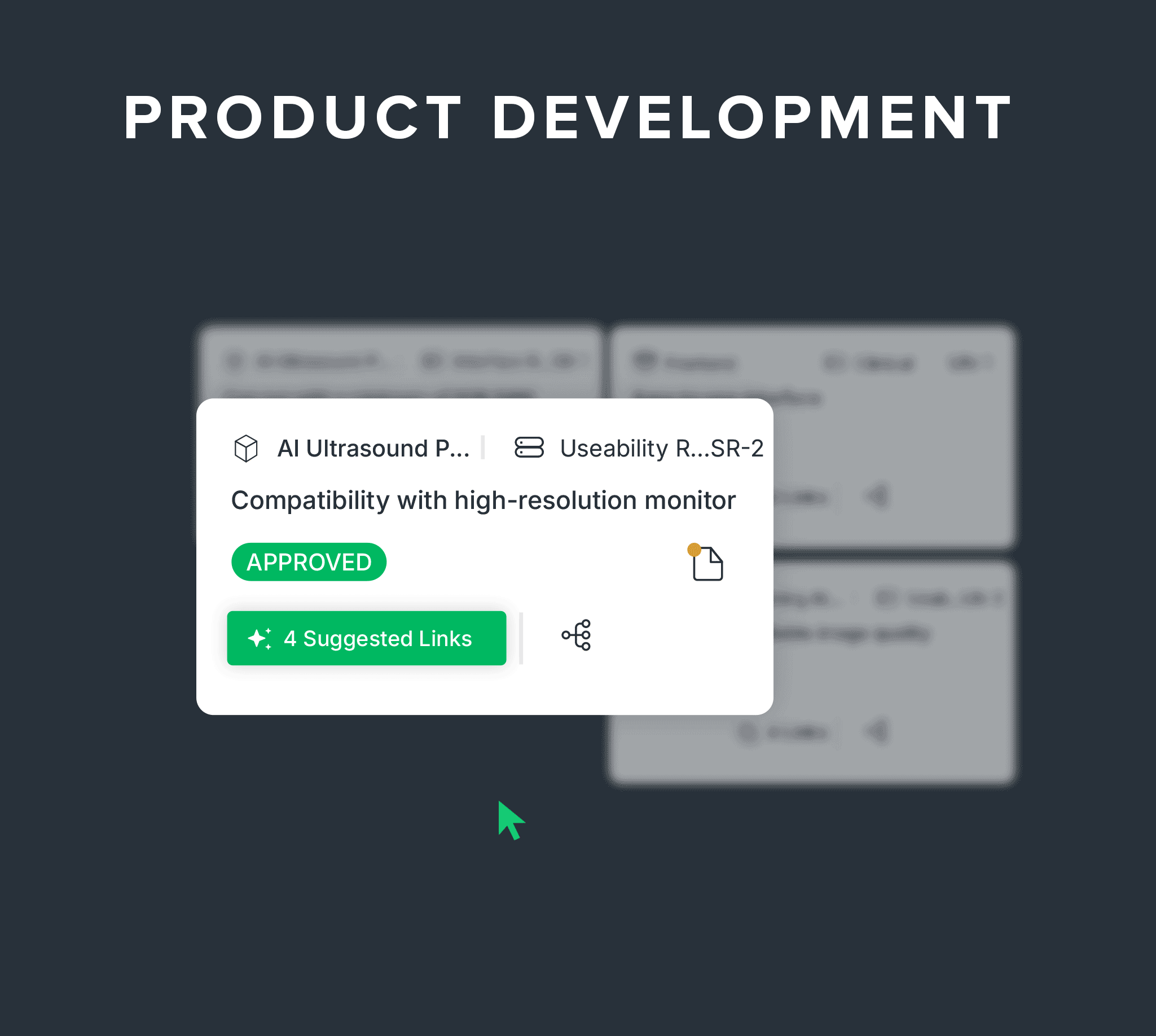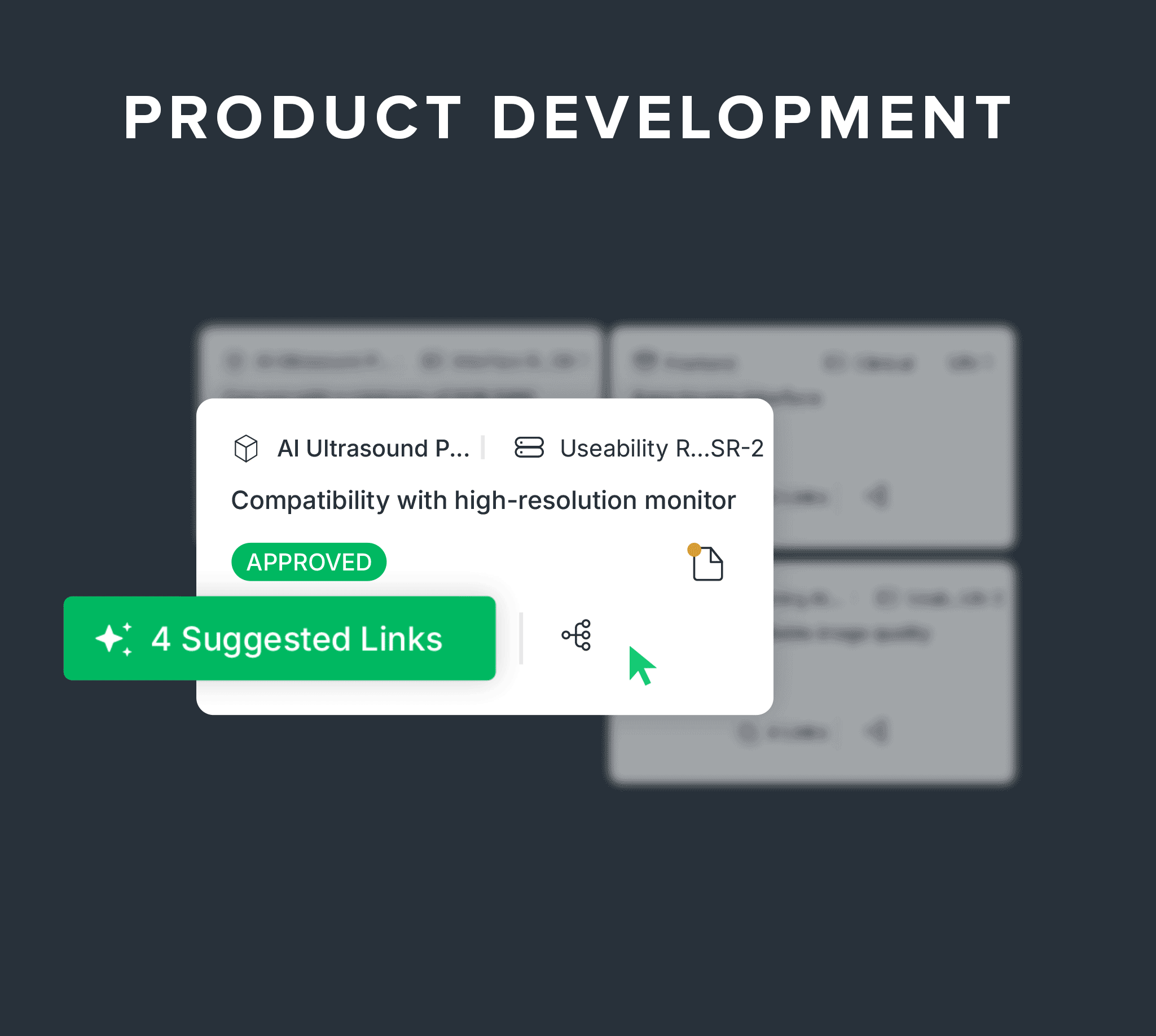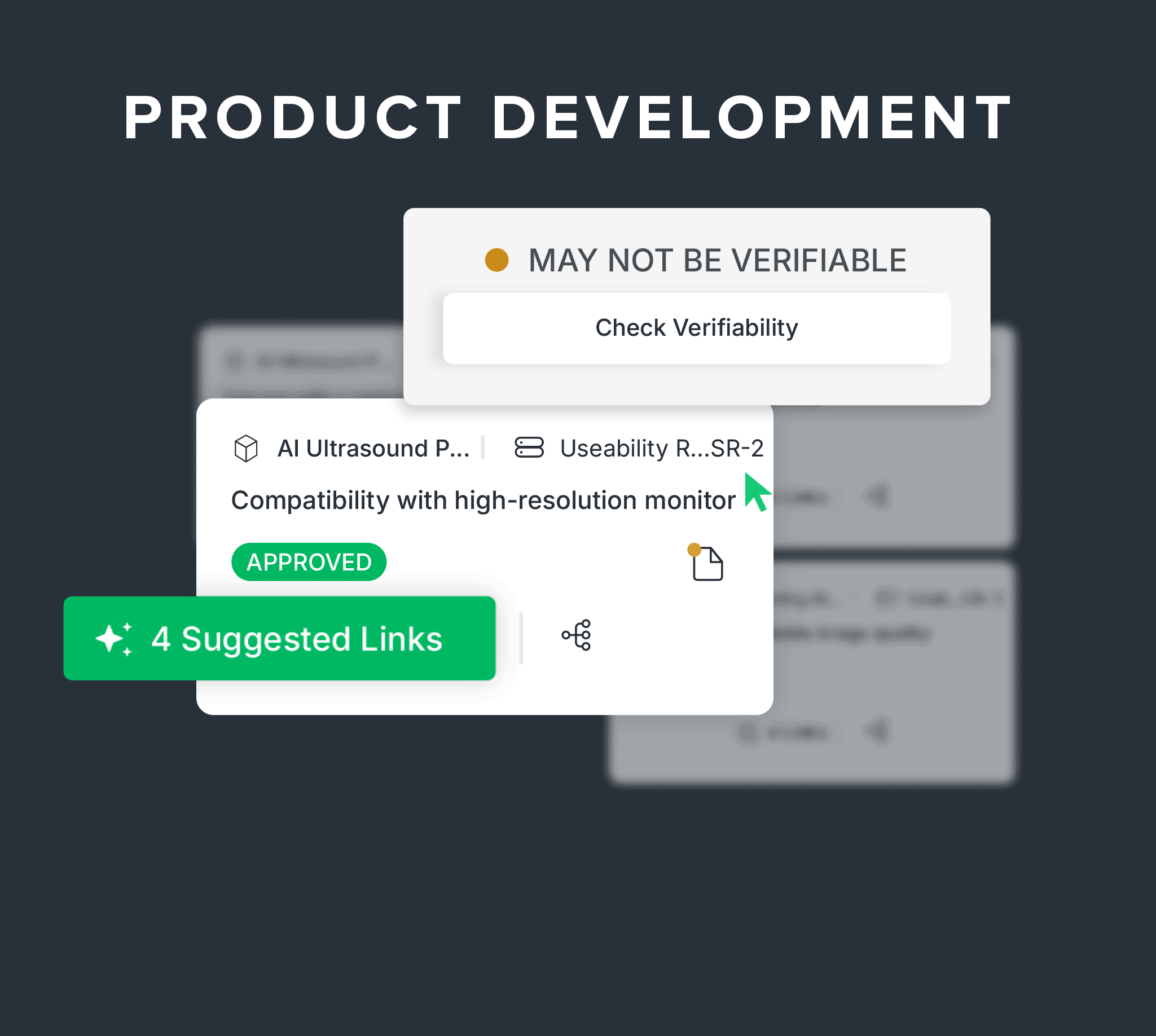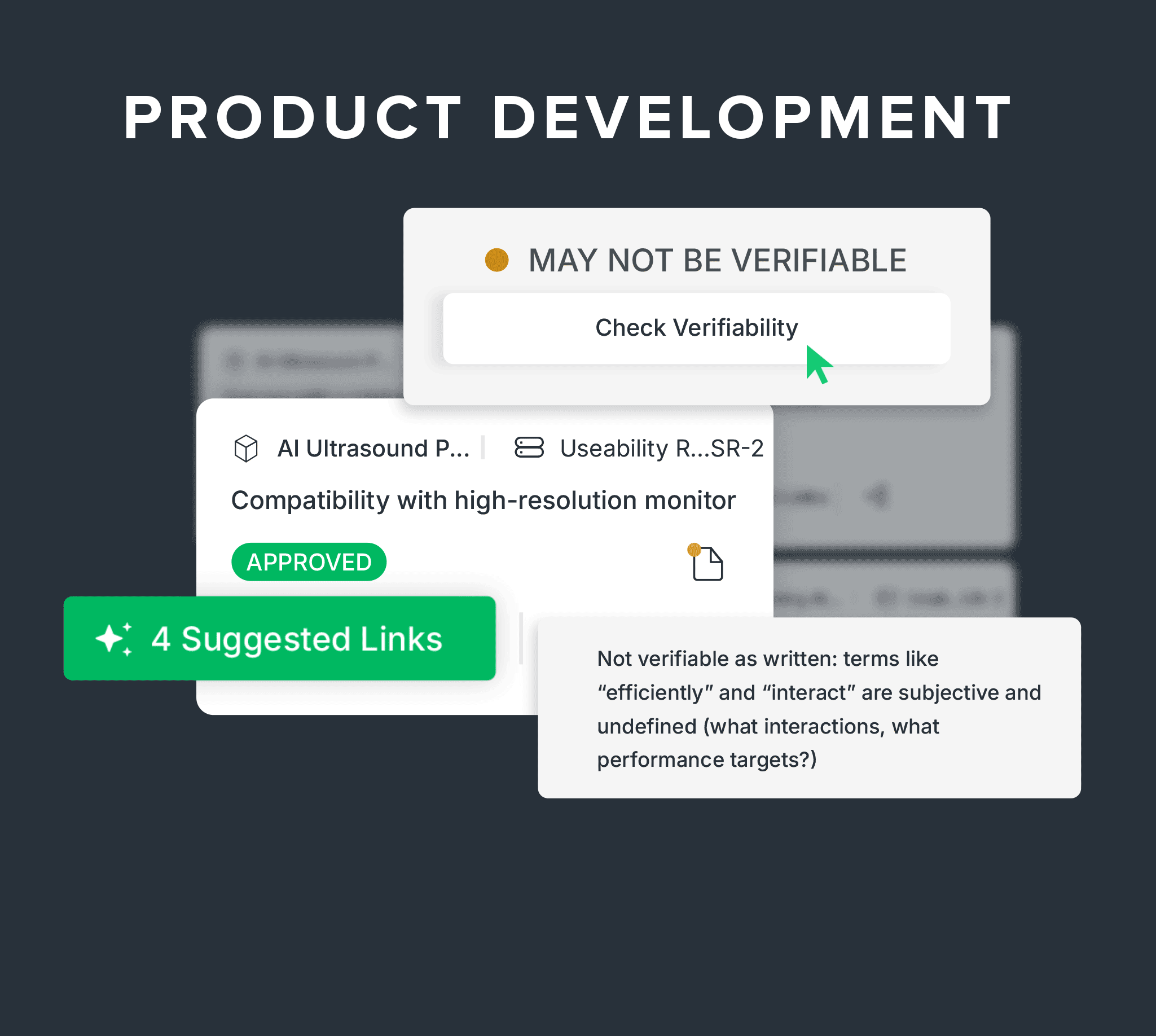
Connect design, risk, and AI-powered traceability as you build.
Link needs, requirements, and verification to design controls.
Automate traceability from dev tools for faster releases.
Assess ISO 14971 risk as requirements and tests evolve.
Oversee studies with connected data, documents, and workflows.
Capture compliant clinical data with a validated medical EDC.
Plan and manage PMCF aligned to EU MDR expectations.
Collect clinical outcome data in-person or remotely on any device.
Built with regulatory insight to help medtech teams move faster with confidence.
Find issues faster and make smarter decisions with less manual work.
Tools and resources that help teams implement quickly and meet requirements.



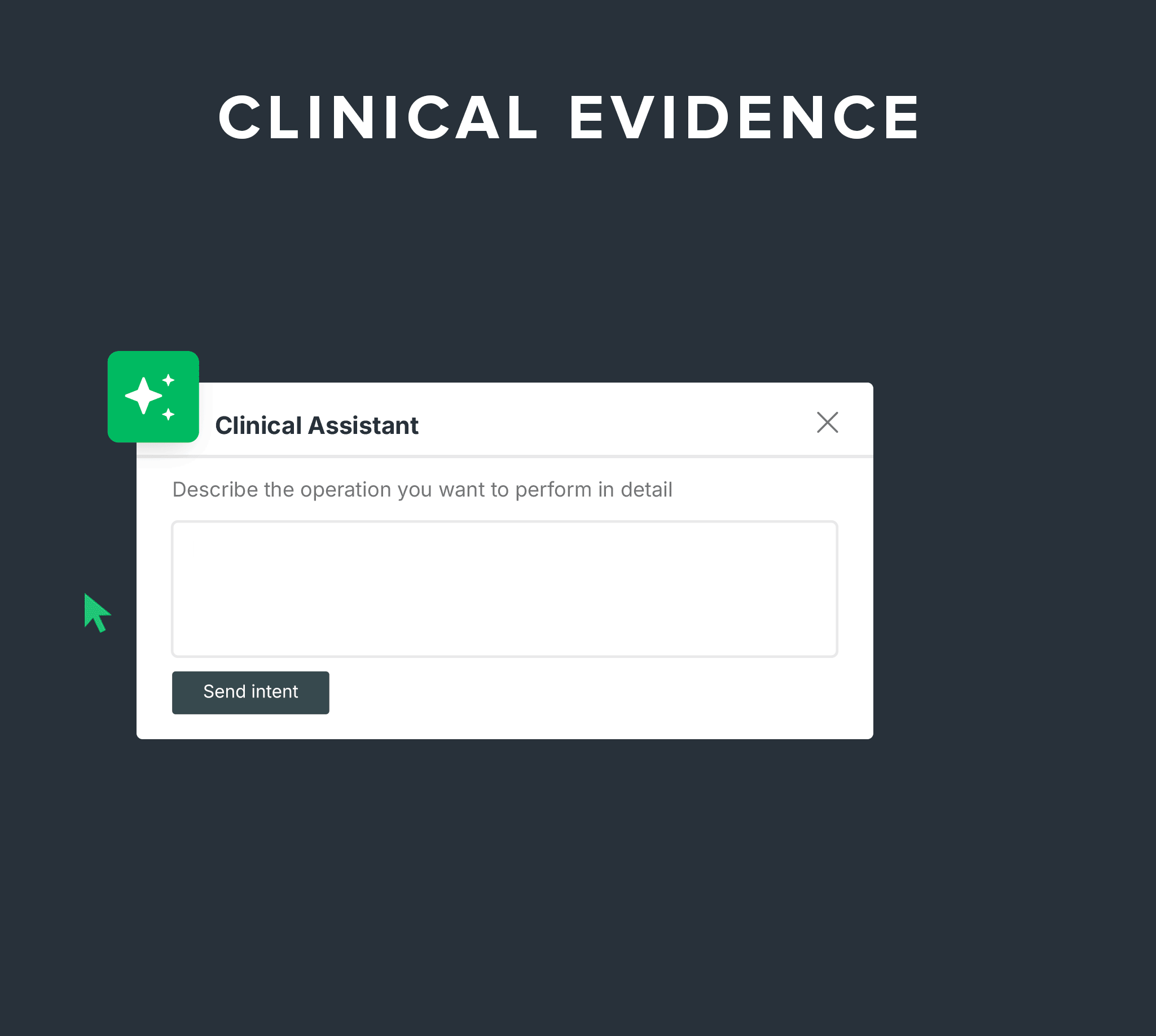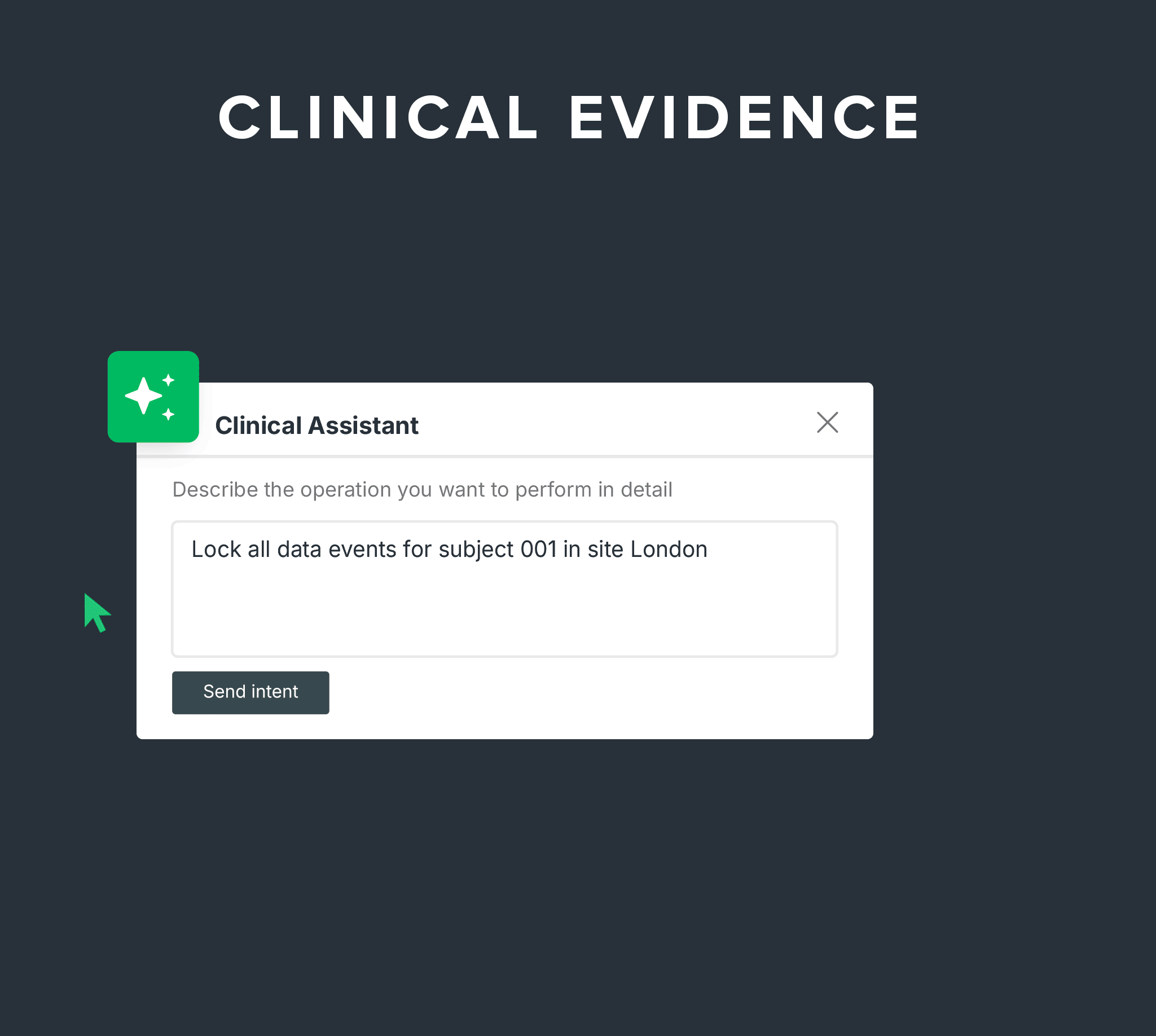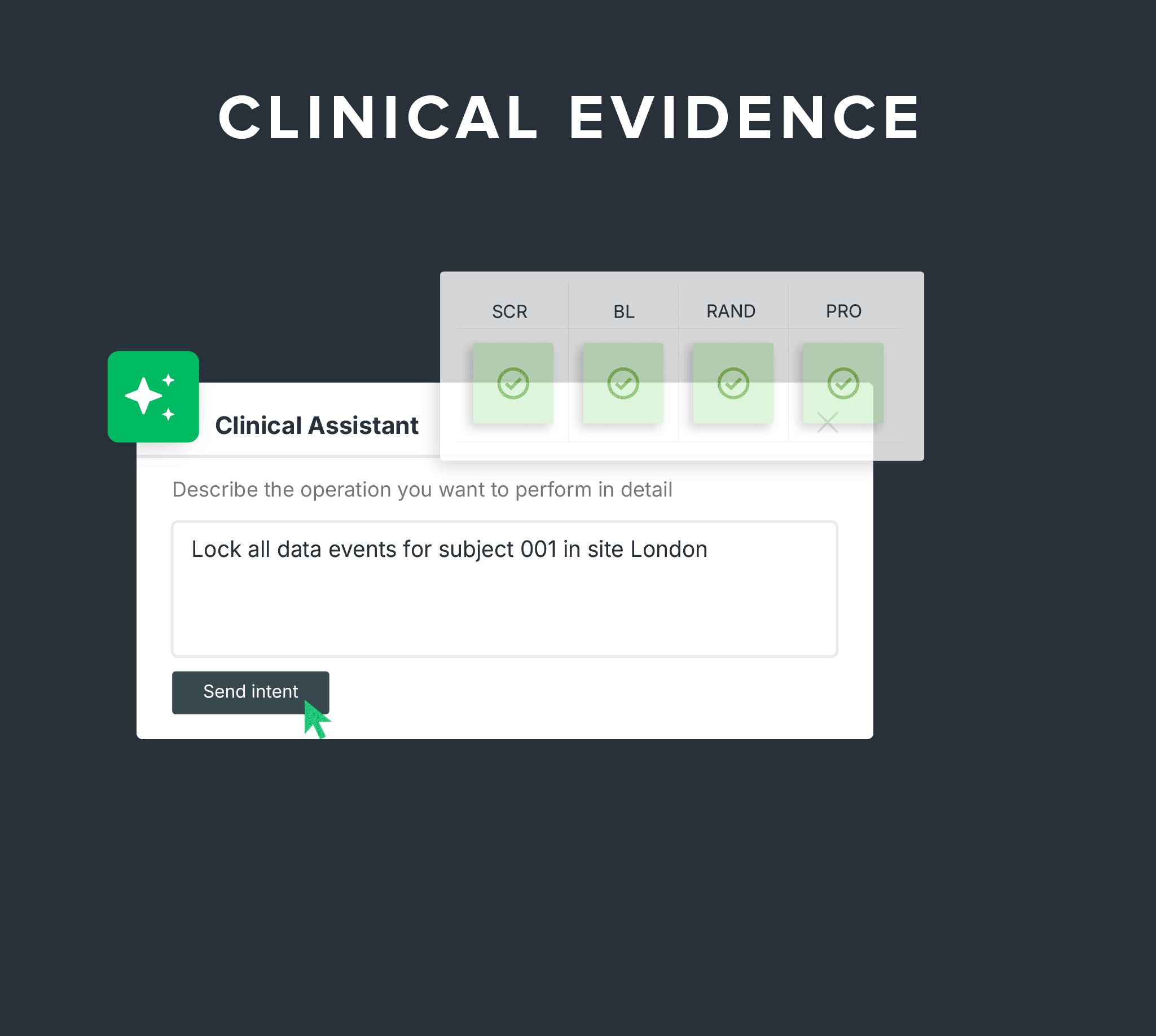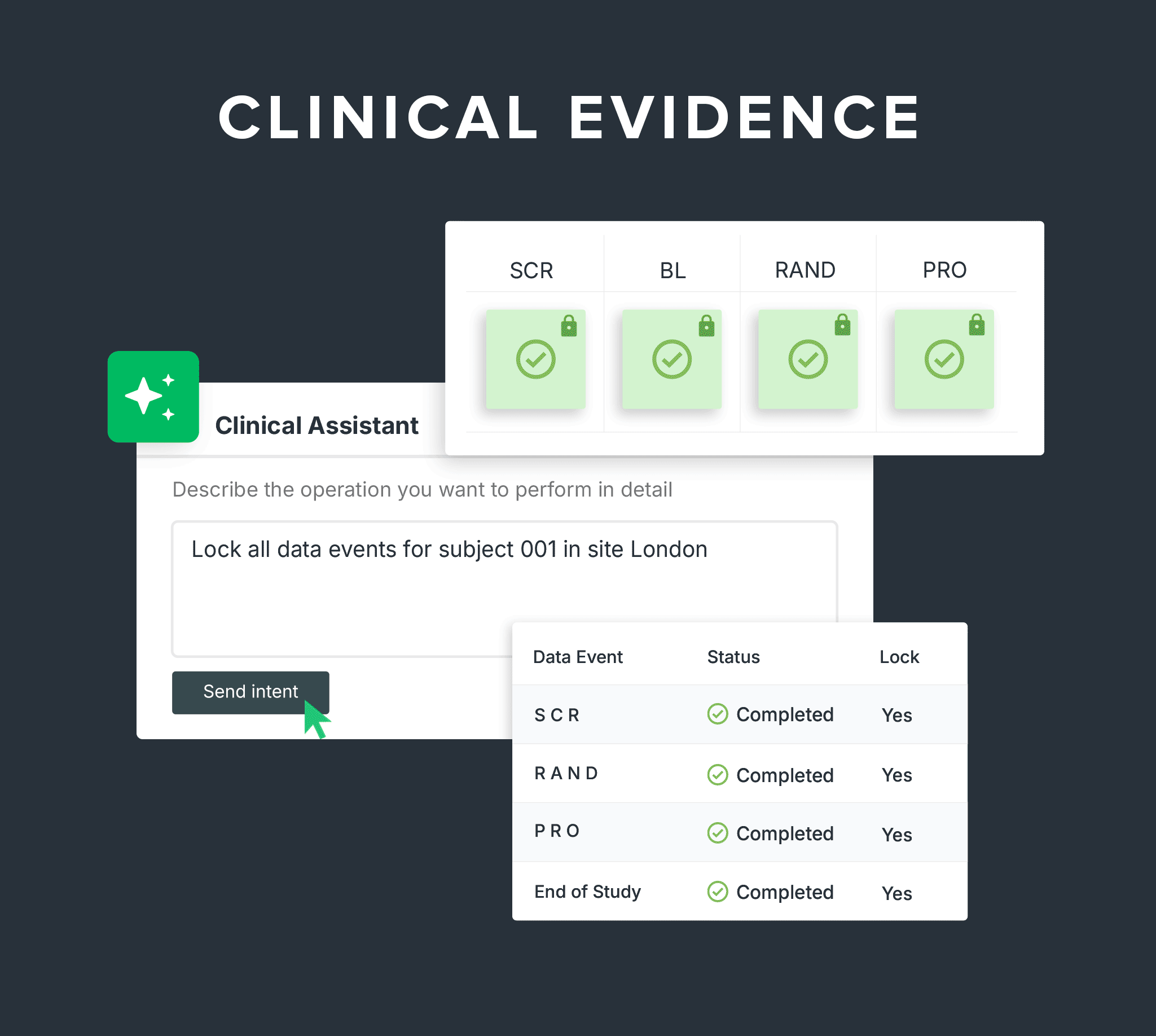
Nobody knows medtech like Greenlight Guru. 1,000+ device companies trust our AI-powered platform to manage quality, product development, and clinical evidence. Spend less time on the system and more time on your device.
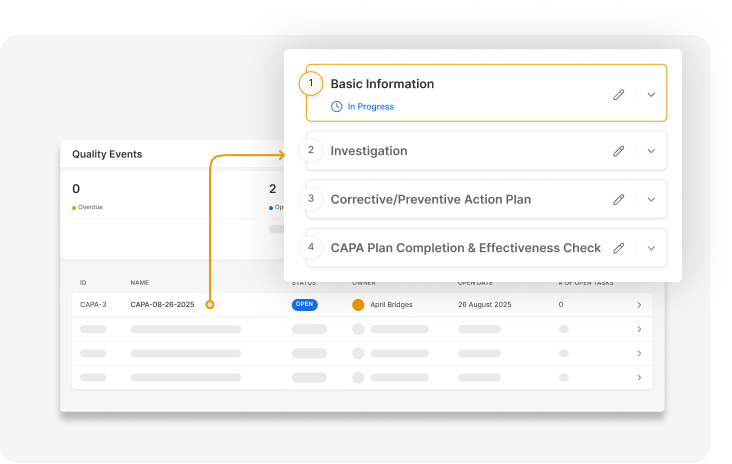
Your quality system stays current and audit-ready without your team manually holding
it together.
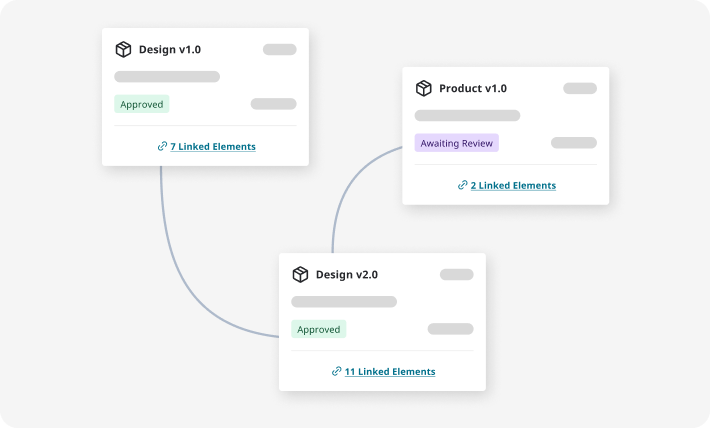
Design controls and traceability that build your compliance record as your
team works.

Run studies, collect data, and own your clinical evidence in one system built for device manufacturers.
Digitally manage your DHF with built-in traceability between design inputs, outputs, verifications, and product risk controls, plus AI-powered suggestions and predictive verifiability checks.

Track and manage operational risk across your QMS – from regulatory requirements to audit readiness and CAPA trends.

Control documents, changes, and training in one place – with automated routing, audit trails, and role-based access.

Log, track, and resolve quality events in a connected system that links CAPAs, training,
and documentation.

Capture compliant, audit-ready clinical data across every phase of your study - from
first-in-human to post-market.



Help your team find answers faster, understand context, and spend less time on manual work without changing how they operate.

Brainstorming
Generate device-specific ideas for design controls and risks so your team can start faster and avoid gaps.
AI Chat
Chat with your QMS data to summarize content, spot patterns, and answer questions without digging through records.
AI-powered global search
Search across your QMS and get direct answers with linked source references for quick validation.
Training analytics
Ask plain-language questions about training completion and get answers back as charts, directly from your data.
EXPLORE THE FEATURES