

.png?format=webp&width=600&height=600&name=applicability-of-iso14155%20(2).png)





.png?format=webp&width=600&height=600&name=plan-clinical-investigation%20(2).png)
Clinical investigations play an important role in your journey of bringing a medical device to market. While they are often perceived as difficult and complex, having a good grasp of the relevant standards can help make the process less confusing.
Understanding ISO 14155:2020 is essential. It is a guide to Good Clinical Practice for clinical investigations of medical devices for human subjects.
This ultimate guide provides an overview of both the standard and Good Clinical Practices (GCP). It further explains important aspects of planning and conducting clinical investigations as per ISO 14155:2020.
(Please note: "Clinical investigation", "clinical trial", and "clinical study" are all synonymous, and I'll be using them interchangeably...)
Table of Contents
 What is ISO 14155?
What is ISO 14155?
ISO 14155:2020 provides the general specifications and requirements for clinical investigations of medical devices. The latest version of the standard was published in 2020 and is intended to serve as a guide for clinical research professionals during the design, conduct, recording, and reporting of clinical trials related to the safety and effectiveness of medical devices.
The purpose of ISO 14155 is to:
- Protect the rights, safety, and well-being of human subjects;
- Ensure scientific conduct of the clinical investigation and credibility of the clinical investigation results;
- Define the responsibilities of the sponsor and principal investigator;
- Assist sponsors, investigators, ethics committees, regulatory authorities, and other bodies involved in the conformity assessment of medical devices.
ISO 14155:2020 is the latest version of the standard. It includes several updates from the previous version.
In the US, FDA recognizes clinical trials that have been run in accordance with ISO 14155, including clinical data collected outside of the US as long as it maintains compliance with the standard.
What's new in the 2020 edition of ISO 14155?
The ISO 14155:2020 and ISO 14155:2011 differ significantly. The 2020 version puts more emphasis on applying risk management principles throughout the entire medical device clinical investigation process.
Annex H of ISO 14155 (Application of ISO 14971 to clinical investigations) explicitly lays out the connection between ISO 14971 and the clinical investigation process. It includes a helpful graphic for applying ISO 14971 to the management of potential safety concerns during a medical device clinical investigation.
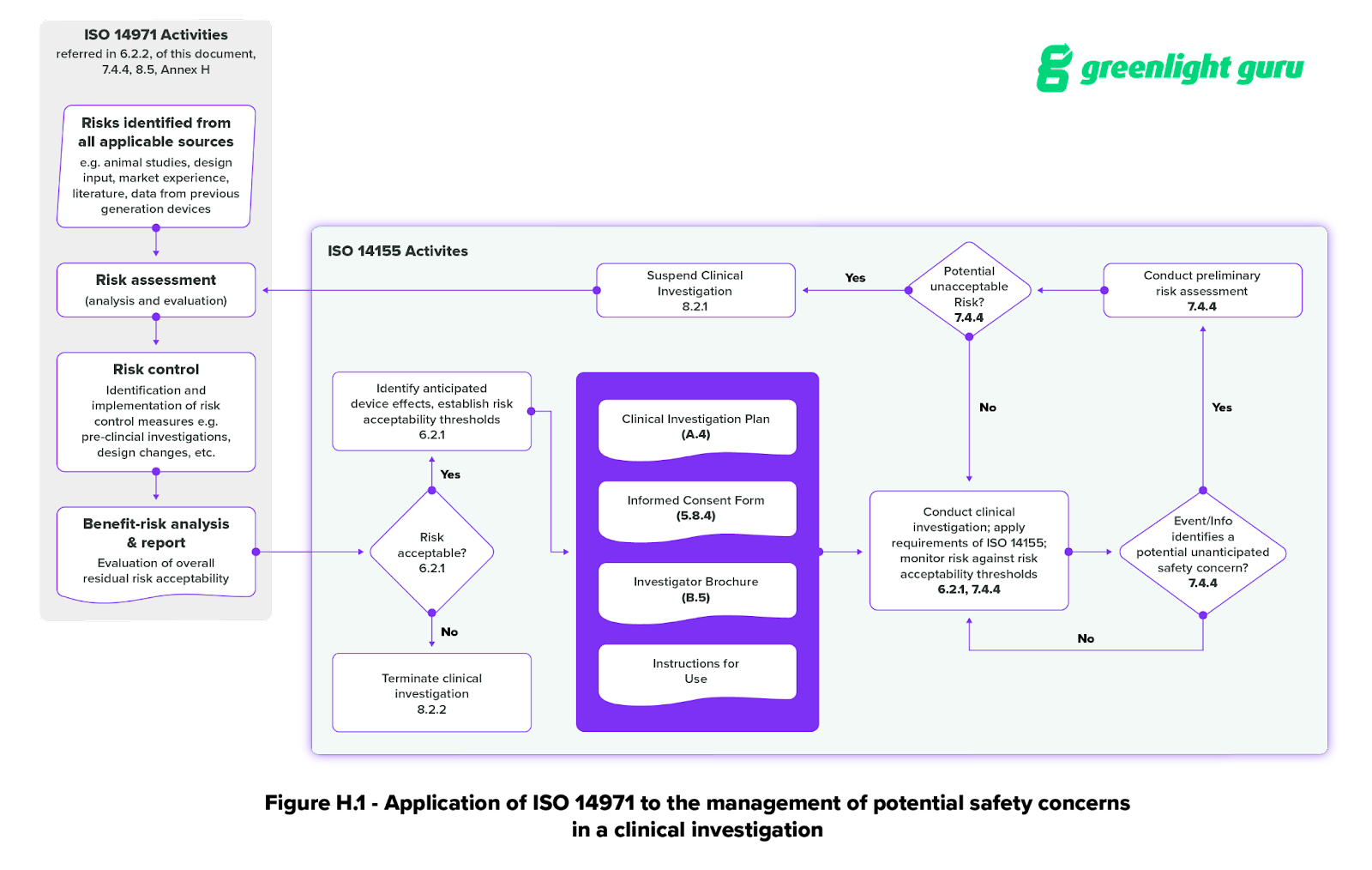
There are other important changes to the 2020 version of the standard, as well, including:
- A summary of Good Clinical Practice (GCP) principles in Section 4
- Requirement for the registration of all medical device clinical investigations in a publicly accessible database like EUDAMED
- The inclusion of clinical quality management in section 9.1
- The inclusion of statistical considerations in Annex A
- Additional guidance for ethics committees in Annex G
- Requirements for risk-based monitoring in Section 6.7
- Guidance on clinical investigation audits in Annex J
- Guidance on the application of ISO 14155 in pre- and post-market stages in Annex I
![how to apply iso14155 in post-market]() How to apply ISO 14155 in post-market clinical activities?
How to apply ISO 14155 in post-market clinical activities?
 How to apply ISO 14155 in post-market clinical activities?
How to apply ISO 14155 in post-market clinical activities?There are numerous types of clinical investigations, and the different language used to describe them can quickly become confusing. On top of that, clinical activities can occur at any stage in the product lifecycle, from pre-market to post-market.
The previous version of the standard did not do a great job of explaining how the standard applied to different types of clinical investigations at different stages in the product lifecycle.
The standard writers have attempted to clear up some of this confusion in ISO 14155:2020 with the addition of Annex I, which clarifies the different types of clinical investigations based on regulatory status, clinical development stage, study design, and burden to subject.
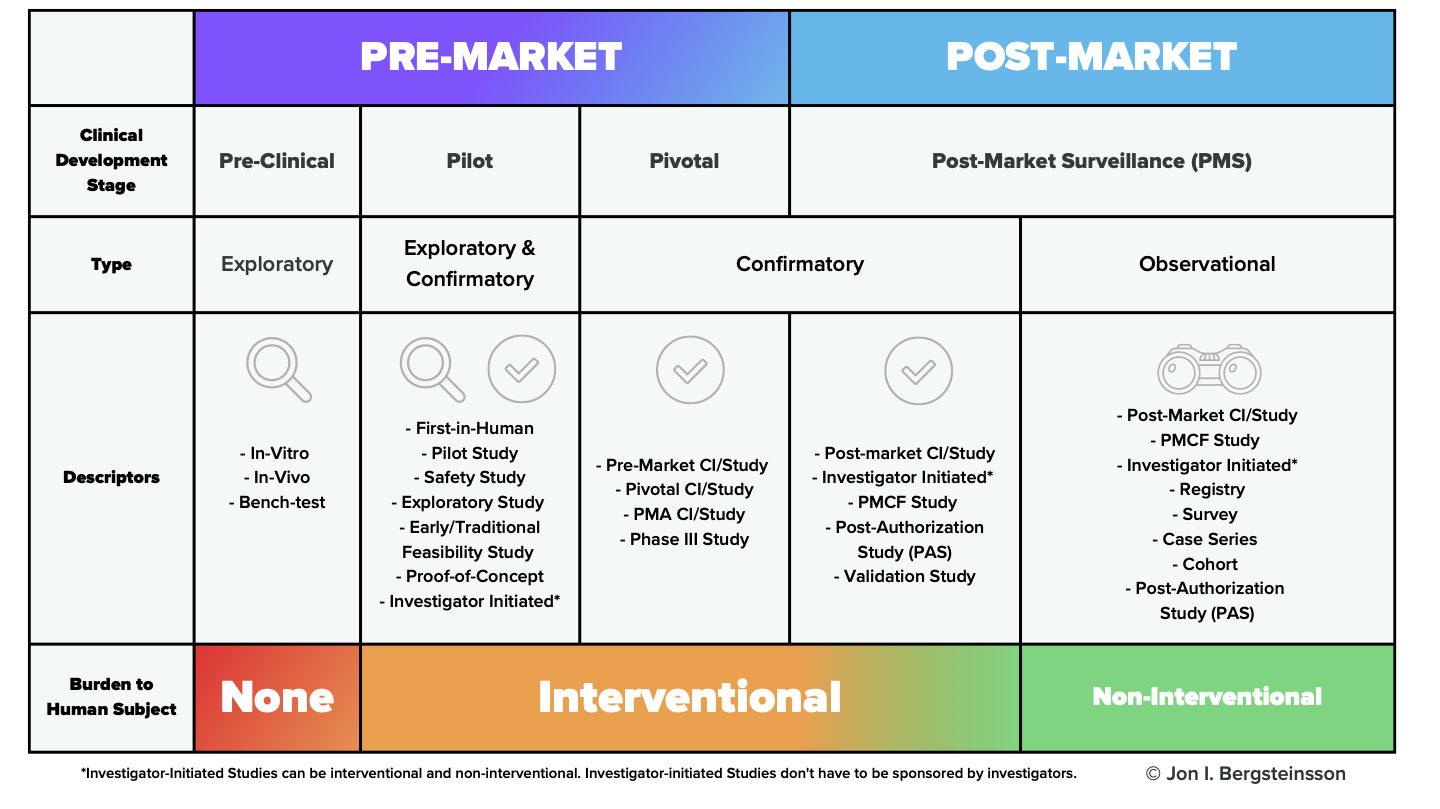
As this image makes clear, clinical investigations of medical devices don't just happen in the pre-market phase. The standard applies to post-market clinical investigations as well, including observational (non-interventional) investigations.
Which parts of ISO 14155 will apply to your clinical study?
In addition to the clarification of the different types of medical device clinical studies, Annex I also includes a section (I.7) on the applicability of the document's principles to different clinical studies.
As the standard states: "Depending on the clinical development stage and the type of clinical investigation design, the principles of this document can be applied in full or in part."
For example, the standard states that for a pre-market confirmatory (interventional) clinical investigation, "all principles in this document apply." On the other hand, if you're undertaking a post-market observational investigation which is non-interventional, you may not be required to apply every requirement in the standard.
The important thing to remember is that if you believe you are exempt from applying any part of this standard to your medical device clinical investigation, you must justify that decision in your clinical investigation plan (CIP).
![what-is-good-clinical-practice gcp]() What is Good Clinical Practice (GCP)?
What is Good Clinical Practice (GCP)?
.png) What is Good Clinical Practice (GCP)?
What is Good Clinical Practice (GCP)?Good Clinical Practice (GCP) is a set of ethical and scientific quality standards. GCP is used for designing, conducting, recording, and reporting trials of medical devices that involve human subjects.
GCP is an internationally recognized standard. It ensures the protection of subjects' rights, safety, and well-being. Additionally, it ensures that all clinical data is credible.
ISO 14155 provides a summary of GCP principles in Section 4 of the standard. These principles are essential for conducting research with human subjects. They form the basis of any good clinical study.
Overall, there are 14 guiding principles of Good Clinical Practice, including:
- All clinical trials should be conducted in accordance with ethical principles, sound scientific evidence and clear detailed protocols
- The benefits of conducting trials should outweigh the risks
- The rights, safety and well-being of trial participants are of paramount importance and these should be preserved by obtaining informed consent and maintaining confidentiality
- The care must be given by qualified personnel with adequate experience
- Records should be easily accessible and retrievable for accurate reporting, verification and interpretation
- Investigational products should be manufactured according to Good Manufacturing Practice
You'll want to familiarize yourself with all the principles laid out in Section 4 of the standard. They are essential to planning and conducting a compliant clinical study for medical devices.
In fact, in the Ethical Considerations portion of the standard (Section 5), the first requirement states that the Good Clinical Practices "shall be understood, observed, and applied at every step in the clinical investigation."
Section 5 also lays out several other important ethical considerations, including:
- Rules against improper influence or inducement of subjects
- Requirements for communication with the ethics committee (EC), including requirements for your initial submission and continuing communication throughout the clinical investigation.
- Requirements for obtaining informed consent and the information that must be provided to the subject. This section is particularly in-depth, as getting informed consent appropriately is critical to conducting an ethical clinical study in accordance with GCP.
.png)
![responsibilities-sponsor-principal-investigator]() What are the key responsibilities of the sponsor and principal investigator during a clinical investigation?
What are the key responsibilities of the sponsor and principal investigator during a clinical investigation?
 What are the key responsibilities of the sponsor and principal investigator during a clinical investigation?
What are the key responsibilities of the sponsor and principal investigator during a clinical investigation?The ISO standard separates the Good Clinical Practices for 'Responsibilities' into two clauses:
- Responsibilities of the Sponsor (Clause 8) and,
- Responsibilities of the Principal Investigator (Clause 9).
The sponsor (in our case, the manufacturer of the device) is responsible for planning and conducting the clinical investigation within prescribed quality assurance and quality control principles.
It's important to note that even if the clinical investigation is contracted out by the sponsor to a qualified third party, the sponsor still retains overall responsibility.
The responsibilities of the Principal Investigator include:
- The implementation and management of the day-to-day activities of the investigation in accordance with the CIP,
- Ensuring the integrity of investigation data,
- Safeguarding the rights, safety and well-being of the human subjects involved in the study.
![how-to-plan-clinical-investigation-iso14155:2020]() How to plan a clinical investigation according to ISO 14155:2020
How to plan a clinical investigation according to ISO 14155:2020
 How to plan a clinical investigation according to ISO 14155:2020
How to plan a clinical investigation according to ISO 14155:2020As you plan your clinical investigation, it's important to understand that a standard like ISO 14155 is not telling you exactly what to do. It's telling you what you need to include and consider as you plan your clinical investigation.
It is important to go through the standard and check off boxes, as well as create the necessary documents. However, this is not the conclusion of your work.
Regulatory authorities will want to see the justification for your decisions.
They'll want to know why you've decided to structure your study a certain way, why you've chosen that sample size, or how you came up with a specific number.
Section 6.3 (Justification for the design of the clinical investigation) has more information on providing that justification.
Keep that in mind as you create the various documents for the planning stage. These documents include:
-
The Clinical Investigation Plan (CIP). The CIP is the go-to document for everyone involved in the clinical study. It will include the objectives and design of your clinical study, as well a justification of the study's design, and a benefit-risk analysis. You can find the full list of what to include in your CIP in Annex A of the standard.
-
The Investigator's Brochure (IB). The purpose of the IB is to provide the principal investigator with sufficient data to justify the clinical investigation proposed in the CIP. You can find the full list of what to include in the IB in Annex B of ISO 14155:2020.
-
Case Report Forms (CRFs). The purpose of CRFs is twofold. Firstly, they provide information on the condition of each subject entering the study. Secondly, they capture data for each subject as required by the CIP.
You can find the full list of what should be included in a CRF in Annex C of the standard.
Now, that basic description of CRFs is deceptively simple. Case report forms are one of the chosen methods of collecting clinical data during a study. Using paper or general-purpose software for your CRFs puts you at risk for missing data or data entry mistakes. These can slow down your study significantly.
At Greenlight Guru, our modern eCRF is built for the unique needs of the MedTech industry. When you use Greenlight Guru Clinical, you can get started very quickly. Ensure peace of mind with our pre-validated software, regulatory templates, and user-friendly study builder.
![how-to-conduct-clinical-investigation-iso14155]() How to conduct a clinical investigation in accordance with ISO 14155:2020
How to conduct a clinical investigation in accordance with ISO 14155:2020
 How to conduct a clinical investigation in accordance with ISO 14155:2020
How to conduct a clinical investigation in accordance with ISO 14155:2020Your clinical investigation may only commence once you have "written approval/favorable opinion" from the ethics committee and the relevant regulatory authority in the countries where your clinical investigation is taking place.
You'll find requirements for conducting an ISO 14155-compliant clinical investigation in Section 7 of the standard. There are a couple points within this section that I want to highlight here.
First, you'll notice that a large part of Section 7 is given over to adverse events and device deficiencies. Documentation and reporting of adverse events or device deficiencies pertaining to medical devices is extremely important. You can find a table for adverse event categorization in Annex F of the ISO standard.
Fortunately, Greenlight Guru Clinical offers an adverse event module for EU MDR and ISO 14155-compliant reporting. The customizable module is fully integrated into our eCRF and provides automatic notifications to users and sponsors and an AE specific data export.
![how-to-manage-risk-iso14155:2020]() Managing risks during a clinical investigation
Managing risks during a clinical investigation
 Managing risks during a clinical investigation
Managing risks during a clinical investigationSection 7 of the ISO standard also touches on risk management, specifically the risk assessment process for potentially unacceptable risks in 7.4.4. This subsection looks at risks that can occur during the clinical investigation. It also includes a written version of the flowchart in Figure H.1.
Section 7 isn't the only place that risk management is mentioned in the standard, however. Section 6.2 provides more risk management requirements for clinical investigations.
Here, the standard states that risk management activities "shall be performed throughout the clinical investigation". And that risk management principles must be applied during both the planning and the conduct of the investigation.
The emphasis here on using ISO 14971 and applying risk management principles throughout the clinical investigation is part of a larger, industry-wide move toward integrating risk management into every part of the medical device lifecycle.
Taking this kind of holistic approach to risk means you may even want to document risk management for clinical investigations within your quality management system. Risk management activities during a Post-Market Clinical Follow-Up investigation may have important implications for the device. These should be documented and tracked in the Quality Management System.
At Greenlight Guru, we understand that risk is a fundamental part of bringing a medical device to market. We built our Risk Management workspace to be traceable and compliant with ISO 14971:2019 and ISO 13485:2016. This ensures our workspace meets the risk-based requirements of both standards.
![electronic-data-capture-compliance-iso14155:2020]() Electronic data capture and compliance with ISO 14155:2020
Electronic data capture and compliance with ISO 14155:2020
 Electronic data capture and compliance with ISO 14155:2020
Electronic data capture and compliance with ISO 14155:2020You'll also want to pay close attention to Section 7.8 (Document and data control). In particular Section 7.8.3, which covers electronic clinical data systems.
If you're using an electronic data capture (EDC) system for clinical data collection during your study, you should know that ISO 14155 requires any electronic system be validated "in order to evaluate the authenticity, accuracy, reliability, and consistent intended performance of the data system."
The standard requires 12 written procedures to be followed and implemented when using any EDC system. These include requirements to:
- Verify and validate that the requirements for the electronic clinical data system can be consistently met
- Ensure attributability, completeness, reliability, consistency, and logic of the data entered
- Ensure that data changes are documented and an audit trail is maintained
- Maintain a security system to prevent unauthorized use of data, both internally and externally
And while that may seem daunting, with Greenlight Guru Clinical, you'll have a system that comes pre-validated to all the requirements in ISO 14155:2020. That includes out-of-the-box compliance with the requirements in Section 7.8.3 and a suite of compliance document templates available to all customers.
For manufacturers at the vendor evaluation stage, our EDC selection guide for device investigations covers the specific ISO 14155 compliance questions to ask every vendor before signing.
![smart-trial-edc-system-for-MedTech]() Final thoughts: Greenlight Guru Clinical is pre-validated for MedTech data collection requirements
Final thoughts: Greenlight Guru Clinical is pre-validated for MedTech data collection requirements
.png) Final thoughts: Greenlight Guru Clinical is pre-validated for MedTech data collection requirements
Final thoughts: Greenlight Guru Clinical is pre-validated for MedTech data collection requirementsPlanning and conducting an ISO 14155-compliant clinical investigation is difficult enough without having to worry about your data collection tools.
That's why we built Greenlight Guru Clinical specifically for the needs of the MedTech industry. Our EDC solution is the leading clinical data collection toolbox for MedTech. It enables you to collect and manage data during pre-market and post-market clinical studies. This includes registries, cohorts, surveys, human factor testing, design validation and more.
Greenlight Guru Clinical meets the regulatory requirements of the FDA, EU, and most other countries. It also ensures out-of-the-box compliance with GCP and ISO 14155:2020.
Get your free demo of Greenlight Guru Clinical today!
FREE eBOOK:
Ultimate Guide To ISO 14155:2020 for Medical Devices
Ultimate Guide To ISO 14155:2020 for Medical Devices

-1.jpg?width=2550&name=Ultimate%20Guide%20to%20ISO%2014155%20(cover)-1.jpg)
-1.jpg?width=250&height=324&name=Ultimate%20Guide%20to%20ISO%2014155%20(cover)-1.jpg)



