
Medical devices are intended to save and improve quality of life.
I like the ISO 13485 definition of a medical device: “Any instrument, apparatus, implement, machine, appliance, implant, in vitro reagent or calibrator, software, material or other similar or related article, intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the specific purpose(s) of:
-
diagnosis, prevention, monitoring, treatment or alleviation of disease
-
diagnosis, monitoring, treatment, alleviation of or compensation for an injury
-
investigation, replacement, modification, or support of the anatomy or of a physiological process
-
supporting or sustaining life
-
control of conception
-
disinfection of medical devices
-
providing information for medical purposes by means of in vitro examination of specimens derived from the human body”
Medical Device Risk Assessment
Medical device risk assessment is the structured way to answer a series of questions: What could harm someone? How bad could it be? How likely is it? And what will we do about it?
In practice, risk assessment covers several steps:
- identify potential hazards and hazardous situations,
- perform risk estimation (severity of harm × probability of occurrence),
- compare results to your acceptable risk criteria, and
- select and verify risk control options.
Under ISO 14971, risk assessment is the combination of your risk analysis and risk evaluation.
Design controls tell you how you’ll design and prove the device works. Risk assessment tells you what must be made safe and how you’ll prove the safety controls work.
Regulatory requirements that drive risk assessment
Risk assessment isn’t optional. Regulators expect it to be built into your development process and maintained after launch.
-
FDA (United States): The Quality Management System Regulation (QMSR) aligns 21 CFR Part 820 more closely with ISO 13485:2016 (quality management systems).
-
EU MDR 2017/745: manufacturers must run risk management as a continuous lifecycle process, including reasonably foreseeable misuse, risk acceptability, and updating controls using post-market information.
-
ISO 14971: the globally recognized framework for the application of risk management to medical devices across the full lifecycle.
-
IEC 62366-1 (usability engineering): focuses on identifying and reducing safety risks related to correct use and use errors.
Intended Use & User Needs
A medical device should address a specific patient and clinical need, defined by manufacturers in an intended use statement. Intended use is key to your product development and risk management efforts, as well as critical to your regulatory strategy, helping to determine classification and go-to-market strategy.
This is really where a medical device’s journey begins. Design controls and risk management should flow and blend together, and it’s important to establish this flow early in product development. Intended use is a gateway to user needs, design & development plan, design inputs, risk management plan, hazards, hazardous situations, and foreseeable sequence of events.
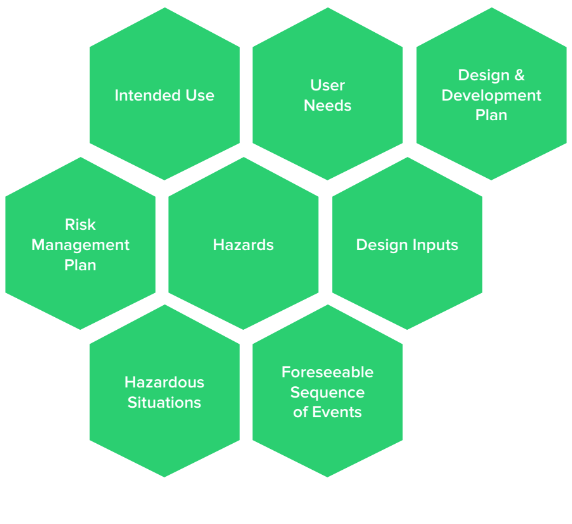
Intended Use Helps Define User Needs
Intended use defines the purpose of a medical device. User needs are then derived and defined from the intended use to describe:
-
Who will use this device?
-
How will the user and patient interact with the device?
-
What type of procedures will the device be used for?
-
What type of environment will the device be used in?
-
When will the device be used?
-
Is the device used one time, or over and over?
-
What other products will the device interact and interface with?
I’ve talked before about the importance of user needs in the design controls process. User needs start the “waterfall,” and design validation closes the process, cycling back to user needs.

Apply Intended Use To Your Design & Development Plan
Intended use and user needs define the scope of a project, and they are instrumental in establishing a design & development plan. Such a plan needs to define:
-
Design & development stages and activities
-
Who is responsible for design & development activities
-
Resources required, such as project team members and key vendors
-
Timing of design reviews
The plan is not a “one and done” and needs to be revisited and updated throughout the project.
Design Inputs Build On User Needs
To define design inputs for your medical device, you need to understand user needs. I’m guessing you can cite several projects where you were able to define design inputs, but there may not have been any defined user needs. Actually, there were user needs —they just might not have been documented, and this can be an issue.
User needs should always be defined and documented. Doing so helps to define design inputs. Otherwise, you might have to guess and fill in the blanks.
When this latter approach is taken, you can progress far into product development before realizing your device has not addressed key aspects from the end-user and patient perspective.
It’s even possible to launch a device into the market and discover later, through product complaints, that you overlooked some user needs.
Intended use usually comprises a few sentences that describe what a product is supposed to do. User needs are a series of statements that further describe the intended use. It‘s okay if your user needs are somewhat vague and ambiguous.
For example, a user need may state something like “the product should be easy to use.” This is where design inputs come into play. They describe and define “easy to use” in clear, objective terms. Think of the design inputs as a contract — a product developer should be able to reference this contract during design and development of the product.
Intended Use Guides Risk Management Planning
One of the first steps involved in medical device risk management is establishing a risk management plan, which describes risk management activities throughout the product lifecycle.
Roles and responsibilities, as well as the risk management team, are defined. Intended use is important because it helps to establish the scope to which risk management activities will be necessary.
The risk management plan also includes criteria for your medical device’s risk acceptability, which should be commensurate with the intended use. Like the design & development plan, a risk management plan is continually evolving throughout the product lifecycle.
Risk assessment steps: the ISO 14971 process, simplified
If you need a simple way to explain the process, use this 7-step flow:
-
Plan risk management (scope, roles, method, and acceptable risk criteria).
-
Identify potential hazards and hazardous situations (including foreseeable misuse).
-
Perform risk estimation (severity x probability of occurrence).
-
Evaluate each risk against acceptability criteria (acceptable / not acceptable).
-
Choose risk control options and implement them.
-
Verify controls work (tests, inspections, validation, usability evidence).
-
Maintain risk management as a living process using production and post-market feedback.
This is where design controls and risk management start to blend: risks drive safety-related design inputs, outputs, and verification activities.
Hazards, Hazardous Situations, Foreseeable Sequence Of Events Based On Intended Use
Risk management also includes risk analysis and risk evaluation — described in Understanding ISO 14971 Medical Device Risk Management. Risk analysis and risk evaluation make up what is known as a risk assessment.

Risk analysis and risk evaluation process aligning with ISO 14971
A risk assessment, based on a device’s intended use, determines the possible hazards, hazardous situations, and foreseeable sequence of events related to your medical device.
A hazard is a potential source of harm, while a hazardous situation is a circumstance in which people, property, or the environment are exposed to one or more hazard(s). The foreseeable sequence of events lays out the steps required for a hazardous situation to result.
Potential hazards: common categories to check
Most devices end up with hazards in predictable buckets. Use this list as a starting point during risk analysis:
- Electrical (shock, leakage, power loss)
- Mechanical (pinch points, breakage, sharp edges)
- Thermal (burns, overheating)
- Biological (biocompatibility, contamination)
- Chemical (toxicity, leachables)
- Software (logic errors, incorrect outputs)
- Use error / human factors (misuse, confusing UI)
- Cybersecurity / data and systems security (unauthorized access, loss of availability)
The goal is not to guess every failure. The goal is to systematically identify hazards tied to intended use and reasonably predictable user behavior.
The Connection Between Design Controls And Risk Management
Just as intended use plays a vital role in design control and risk management, design control and risk management are vital to one another. Some companies treat design controls and risk management as related but separate processes, not realizing the close connection between user needs, design inputs, hazards, and hazardous situations.
Intended use leads to user needs, which lead to design inputs — the “contract” by which medical devices are designed and developed. Intended use also helps define the scope of a design & development plan, as well as the scope of a risk management plan. All of this information is used to determine hazards, hazardous situations, and foreseeable sequence of events.
As I stated at the beginning of this article, medical devices are intended to save and improve quality of life. Following sound design controls and risk management processes helps ensure that the devices you design, develop, manufacture, and sell are as safe and effective as possible.
Verification, Validation & Risk Controls
Part 1 of this series addressed design control and risk management connections through intended use and user needs — specifically, how these items are key to identifying hazards, hazardous situations, and foreseeable sequence of events.
To recap, intended use leads to user needs, which lead to design inputs. Think of design inputs as a “contract” by which medical devices are designed and developed. Intended use also helps define the scope of a design & development plan, as well as the scope of a risk management plan.
Let me also remind you of the high-level risk management process overview defined in ISO 14971:
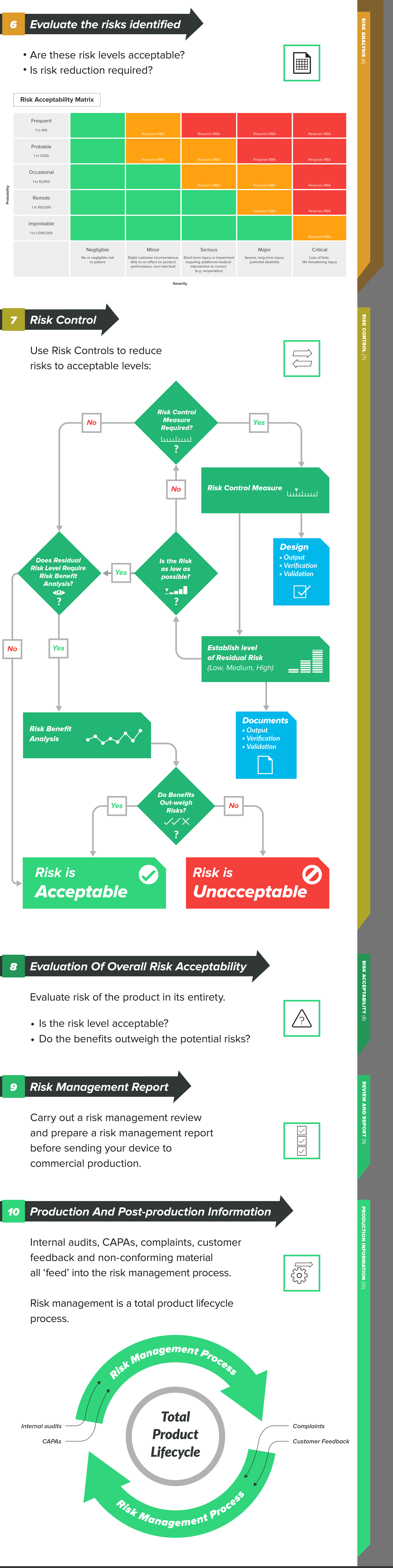
Here, I’ll continue the journey of demonstrating how design controls and risk management should flow seamlessly back and forth to improve safety and reduce risks associated with medical devices, tying in the connections that design inputs, design outputs, design verification, and design validation have with risk controls.
From the perspective of risk management, you have estimated the risks of each hazardous situation by determining severity of potential harm and its probability of occurrence.
You established risk acceptability criteria earlier, during the risk management planning phase of product development. Chances are, you have some type of risk acceptability matrix or chart where you have defined regions of both acceptable and unacceptable risks.

Based on the risk acceptability criteria you have established, there is one significant question you need to answer at this stage: Is risk reduction necessary? If you have made the design controls and risk management connection, then the answer to this question will be a driving force for the next several stages of your medical device product development.
The risk control “phase”
When you have identified hazardous situations requiring risk reduction, you enter the risk control phase of the risk management process.
According to ISO 14971, risk control is the process in which decisions are made and measures implemented by which risks are reduced to, or maintained within, specified levels.
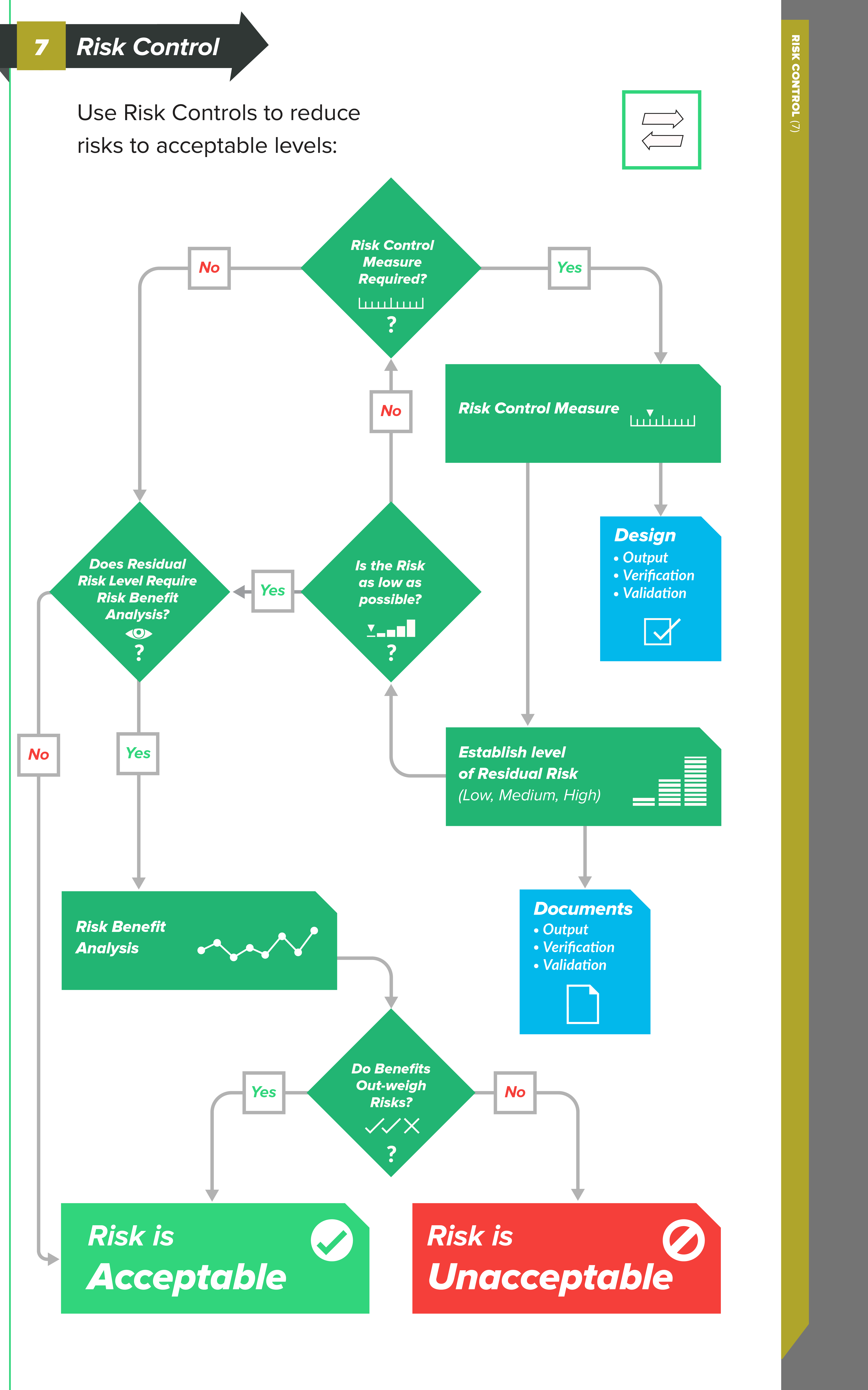
Types of Risk Control measures
In order to reduce risks to acceptable levels, you need to identify possible options — risk control measures — that are appropriate and substantial. There are three basic types, or levels, of risk controls you should consider, listed here in order of priority:
-
Inherent safety by design — Ensure that the design of the medical device reduces and/or eliminates the probability of harmful occurrences altogether.
-
Protective measures in the medical device and/or manufacturing process – Examples of this could include redundant features, safety mechanisms, etc., with the intent to reduce the occurrence of harm.
-
Information for safety – This category of risk controls — which includes labeling, instructions for use, training materials, and the like — is largely regarded as least effective, so much so that ISO 14971 does not allow you to use this as a means for risk control because information for safety is a general requirement of medical devices.
Risk control options
When a risk is not acceptable, you choose risk control options using this hierarchy:
-
Inherent safety by design (remove the hazard or reduce it through design)
-
Protective measures (guards, alarms, interlocks, manufacturing controls)
-
Information for safety (labeling, warnings, IFU, training)
This priority order is also reflected in EU MDR Annex I language on reducing risk through design first, then protections, then information.
After controls are implemented, you re-estimate risk and document the residual risk (what remains).
The connection between Risk Controls and Design Controls
As you can see, there is a strong connection between the levels of risk control measures and design controls.
When you identify risk control measures, these items can drive changes and revisions to your design inputs. Perhaps your design inputs can be better clarified and stated. Or, perhaps you need to include additional design inputs.
Design outputs, too, can be significantly influenced by your risk control measures. Remember that the design outputs established during medical device product development serve as the preliminary device master record (DMR) for the product.
In other words, the design outputs are the “recipe” for your medical device. Risk control measures relating to design outputs could lead to adding new safety features into your product and providing more clarity and definition in design output documents.
KEY TIP: I like to do a first pass through risk analysis, risk evaluation, and risk controls after I have my user needs and design inputs defined, and BEFORE doing too much work on my design outputs, design verification, and design validation. Why? I want to know what my initial risk levels are before creating overly complicated drawings and specifications, and before conducting expensive — and maybe unnecessary — verification and validation (V&V) testing.
Identifying risk control measures should help you define what type of design verification activities will be necessary to demonstrate that your design outputs meet your design inputs, and to prove your medical device is safe. Design verification activities are also instrumental in providing some objective evidence to support probability of events that could lead to harm.
As with design verification, risk control measures can also help shape what type of design validation will be necessary to prove the product addresses user needs.
Design validation can serve as a means to determine if hazards and hazardous situations are likely during intended use. Results of design verification and design validation will be influential when estimating and evaluating residual risks.
Residual Risk Acceptability
After identifying all risk control measures and implementing them, the next step is to evaluate and estimate the residual risks: Did you successfully reduce risks to acceptable levels? If not, consider additional risk controls.
Or, maybe you can conduct a risk/benefit analysis where you evaluate and weigh the medical benefits your device provides against the risks. If you choose this path, document the explanation. Did your risk controls introduce new hazards and hazardous situations? If so, analyze, evaluate, and estimate the risks from these new scenarios.
Risk control measures are the key to identifying ways to mitigate and reduce your product’s risks to acceptable levels. Risk controls provide a means to help you develop your medical device “recipe” through design outputs, to prove these outputs meet design inputs via design verifications, and to prove your medical device meets end user needs.
As I have stated before, medical devices are intended to save and improve quality of life. Following sound design controls and risk management processes helps ensure that the devices you design, develop, manufacture, and sell are as safe and effective as possible.
Using Design Reviews Effectively
Design controls and risk management are key to the success of a medical device, in that they demonstrate that your product is safe and effective for its intended uses. Furthermore, there is a strong, complementary relationship between design controls and risk management, something we have discussed in this series’ previous installments.
Part 1 explored design control and risk management connections through intended use and user needs, while part 2 connected risk controls to design outputs, design verification, and design validation. In this installment, I’ll discuss best practices regarding design reviews, and how to incorporate risk management as a critical element helping to drive decisions.
Design Reviews — Breaking Down The Requirements
I like to start with the good ol’ design controls “waterfall” diagram to show how design reviews fit into the medical device product development paradigm:
Design reviews are intended to encompass all aspects of design controls. Yes, that’s correct —all design control efforts should be included as part of a design review throughout the product development process.
Consider what FDA 21 CFR part 820.30(e) and ISO 13485:2016 section 7.3.5 state about design reviews:
820.30(e) - Design review:
Each manufacturer shall establish and maintain procedures to ensure that formal documented reviews of the design results are planned and conducted at appropriate stages of the device's design development. The procedures shall ensure that participants at each design review include representatives of all functions concerned with the design stage being reviewed and an individual(s) who does not have direct responsibility for the design stage being reviewed, as well as any specialists needed. The results of a design review, including identification of the design, the date, and the individual(s) performing the review, shall be documented in the design history file (the DHF).
ISO 13485:2016 - section 7.3.5 - Design and development review:
At suitable stages, systematic reviews of design and development shall be performed in accordance with planned and documented arrangements to:
a) evaluate the ability of the results of design and development to meet requirements;
b) to identify and propose necessary actions.
Participants in such reviews shall include representatives of functions concerned with the design and development stage being reviewed, as well as other specialist personnel.
Records of the results of the reviews and any necessary actions shall be maintained and include the identification of the design under review, the participants involved and the date of the review (see 4.2.5).
The key take-home points from both FDA and ISO for design reviews are that they must:
-
Be planned at suitable and appropriate stages during product development
-
Include applicable functions for the stage being reviewed
-
Include an “independent reviewer”
-
Include documented records of the events
Design Review vs. Phase Review
Taken literally, the waterfall diagram suggests that design controls and subsequent design reviews follow a serial, or linear, progression.
Use of this methodology to manage product development projects is common in, but not unique to, the medical device industry. The “phase-gate model” (branded as Stage-Gate by Drs. Robert Cooper and Scott Edgett) bears a striking resemblance to the design control stages identified in the waterfall.
The basic premise is that each phase, or stage, is defined with minimum criteria and milestones. Before moving to the next phase, you need to satisfy that the criteria have been met, and the way to do so is via formal phase review.
I bring this up for one primary reason. The medical device design control process is more or less a phase gate approach, but with a slight twist.
The purpose of a phase review is to make a business decision — whether or not to continue funding a project, with dollars and resources, into the next phase of product development.
Design reviews serve a similar purpose, but dollars and resources are not the key metrics to monitor. With a medical device, the purpose at each stage is to demonstrate an acceptable level of safety and efficacy before continuing to the next stage. Design reviews are a mechanism used to assess and document these decisions.
It’s worth noting that there are ongoing debates on whether design reviews and phase reviews both are required for medical device product development.
One point of view suggests that these should be separate events: Phase reviews analyze business decisions; Design reviews analyze design controls. The other point of view suggests that blending phase reviews with design reviews is perfectly fine.
When you’re deciding whether to separate or combine these events, ask yourself—who needs to be present for this review? If it’s purely technical, it may make more sense for your organization to break these meetings apart and have separate Design Reviews. If you do decide to combine phase reviews and design reviews, do yourself a favor and capture the design controls details on a design review form and other non-design control items in separate notes. We all have too many meetings as it is. Try to simplify things a bit when you can.
Must Medical Device Product Development Be Linear?
In a word: No.
Conventional wisdom suggests that linear product development processes are generally less risky (in a project sense of the word), versus processes that allow parallel activities. The en vogue product development practices involve lean and agile approaches, though, and you definitely can utilize such approaches in medical device product development.
But there are a few medical device design control absolutes that must occur and be documented:
-
A design output must include or make reference to acceptance criteria.
-
Design verification must demonstrate that design outputs meet design inputs. A design verification can only be conducted after design output / design input relationships have been established.
-
Design validation must demonstrate that a product meets user needs. A design validation can only be conducted after product and user needs are defined.
-
Design validation requires products to utilize production processes (or equivalents).
-
All design controls must be part of design review(s).
Linking Risk Management To Design Reviews
Again, the primary purpose of both risk management and design controls is to ensure that your product is safe and effective for its intended uses. Demonstrating this requires objective evidence documented in a design history file and a risk management file.
Blending design controls and risk management, rather than treating them as entirely independent workflows, will improve your medical devices, and one way to bring these practices together is via design reviews.
As noted, you need to conduct design reviews at appropriate stages during design and development. I’ll let you decide on how often and how many.
But since you are asking, my advice is to conduct a minimum of five design reviews, one at each of these stages: user needs, design inputs, design outputs, design verification, and design validation.
Why? You need to show with objective evidence (i.e., documentation) that all design controls have been part of design reviews. The simplest and cleanest way to do so is by having separate design reviews for each of the major design control elements.
And, at each and every one of your design reviews, risk management should be the centerpiece of the event. Use risk management as a tool, rather than a checkbox activity.
Use your ISO 14971-compliant approaches to drive risk-based decision-making as a practice within your medical device product development efforts.
Identify where your product risks are, and discuss how to mitigate and control those risks via design controls; document these discussions as part of design reviews.
Understanding how to blend design controls and risk management efforts into a continuous stream will bring purpose and meaning to your product development efforts.
As regulatory bodies around the world harmonize their concepts of “risk-based approaches,” and tout the importance of complying with ISO 14971, you must evolve your own internal practices, as well.
Quality management systems: keeping risk management connected and current
A quality management system (QMS) should make risk management a living process—not a one-time spreadsheet exercise. The FDA’s QMSR aligns more closely with ISO 13485 concepts, which reinforces the expectation that risk management stays active across the lifecycle.
Risk assessment documentation
A typical Risk Management File includes:
-
Risk management plan (scope, roles, methods, acceptable risk criteria)
-
Hazard analysis (hazards, hazardous situations, foreseeable misuse)
-
Risk estimation + risk evaluation outputs (risk matrix or equivalent)
-
Selected risk controls and verification evidence
-
Residual risk evaluation and, when needed, benefit-risk rationale
-
Post-market inputs and updates (complaints, CAPA, surveillance)
Greenlight Guru’s Risk Solutions is aligned with ISO 14971:2019 and risk-based requirements of ISO 13485:2016
Making the connection between risk management and design controls is an essential part of bringing the safest, most effective medical devices to market.
But if you’re using software that doesn’t facilitate those connections—generic solutions that aren’t built for MedTech—then it’s going to be difficult to control the risks associated with your device’s design.
At Greenlight Guru, all of our products are purpose-built for medical device companies just like yours. Our comprehensive risk management software, Risk Solutions, was built with your needs in mind. That’s why Risk Solutions is aligned with ISO 14971:2019 and the risk-based requirements in ISO 13485:2016.
So if you’re ready for risk management software that’s built specifically for you, then get your demo of Greenlight Guru today!
Looking for a design control solution to help you bring safer medical devices to market faster with less risk? Click here to take a quick tour of Greenlight Guru's Medical Device QMS software →
FREE eBOOK:
Risk Management + Design Controls Connection: What Device Makers Need to Know
Risk Management + Design Controls Connection: What Device Makers Need to Know

%20Risk%20Management%20+%20Design%20Controls%20Connection_%20What%20Device%20Makers%20Need%20to%20Know.png?width=5100&name=(cover)%20Risk%20Management%20+%20Design%20Controls%20Connection_%20What%20Device%20Makers%20Need%20to%20Know.png)
%20Risk%20Management%20+%20Design%20Controls%20Connection_%20What%20Device%20Makers%20Need%20to%20Know.png?width=250&name=(cover)%20Risk%20Management%20+%20Design%20Controls%20Connection_%20What%20Device%20Makers%20Need%20to%20Know.png)



