





Table of Contents
|
Design and Development Planning Build Supplier and Partner Network |
Ideas for medical devices can come from anyone at any time. Experienced engineers from large companies and highly recognized surgeons are not the only people capable of bringing a novel device to the market. You might be sitting at a doctor’s appointment thinking “There has to be a better way to check this.” Or, you might be taking care of an ailing family member and want something to assist them in their recovery process.
For those of us who don’t work for large companies, the idea often stops right there because the medical device industry is thought of as being too regulated and having too many roadblocks. While it is true that there is a lot to consider, and it can be an intimidating process, it is also a very rewarding one that is well worth pursuing.
Many of those who have decided to take the plunge find themselves asking a simple question: “Where do I start?”
If you are like me, you have spent countless hours searching the internet, hoping to find a “one size fits all” guide to help you answer that question and many others you may have. Of course, there is no one, single guide that will define the exact path you need to take for your specific device. There are ways, however, to simplify the processes involved in order to create a manageable plan that works for you.
To assist you in these efforts, I’ve created this step-by-step guide to bringing a medical device to market. The goal is to give you a go-to guide that is easy to follow, can point you in the right direction, and will ultimately set you up for a successful product launch. So, let’s get started...

![UG-BMDGM_building biz case_1]() Building The Business Case
Building The Business Case

Before even thinking about interacting with the FDA or your notified body, you must first make sure there is a business case for your device. Many people want to rush to the “fun part” of building and testing their product, but it is very important to put in the necessary work upfront to develop a solid business plan.
Ideation
Everything begins with an idea.
Before you can come up with a solution, you need a problem to solve. Maybe you’re starting from scratch to tackle a medical issue that has no current solution. Or, you might be attempting to make existing technology more efficient and/or affordable.
While it is okay to shift your focus as your project progresses, it’s vital to start with a clear idea of the problem you’re trying to solve. Consider medical conditions and current treatment options, user needs, and existing technology, and go from there.
This work does not have to be done all on your own. Make sure you identify and connect with all of the different stakeholders early in the process.
Doctors, nurses, and other medical professionals are great resources when brainstorming ideas. They live on the front lines and will be able to give you a personalized perspective of the pluses and minuses of any existing products that may be similar to yours. They may even tell you that there is not a single device on the market like the one you’re looking to develop. This information can be helpful in gauging potential market demand, as well.
Patients, such as friends or family members, can tell you about unpleasant experiences they may have had while being treated using a certain existing device. Their personal stories can be especially powerful in motivating you to bring your better solution to life.
Sales professionals are often overlooked, but they have great ideas and can provide helpful feedback about how devices are actually used in their real setting, as well as potential areas for improvement. After all, these people are the ones receiving the initial feedback directly from the end users.
Taking inspiration from others, including colleagues, end-users, and even friends and family can exponentially help you to improve and solidify your idea.
Market Research
Having a great idea for a product isn’t enough to guarantee its success in the market. You’ll first need to make sure there is in fact a market for your product. I strongly recommend researching any potential competitors to measure your proposal up against their existing products.
Here are some questions that every aspiring device developer should ask themselves:
- Will my device fill an unmet need?
- Who would buy this device, and where in the world could I sell it?
- What are the competitive products / technologies / services currently available?
I recommend that you take the time to write out a description of your product, and then make a list of the answers to these questions. This list doesn’t need to be set in stone, but it’ll serve as a good “back-of-the-envelope” resource for you to have as you determine market demand.
This list is also especially important because the answers to the questions will help you outline the amount of time and capital your project is going to take.
Market research is key - different countries and regions will pay different prices for the same device, but it’s not just about crunching numbers. Cultural research is one major factor to keep in mind - certain cultures may be biased toward or against certain types of medical procedures or devices.
The size of your target market is important for a number of reasons. If a target market is too small, you will want to determine early on that it’s not worth pursuing.
Market size will also likely dictate how much capital you can raise if you decide to go that route. While it’s possible to determine the size of certain individual markets based on existing data, international markets are more difficult to gauge.
A good rule of thumb is to assume that an international market is around the size of the US market.
Groups like IMS Global and Frost & Sullivan have published market studies for the medical device industry.
When it comes to measuring yourself against competitors, ask yourself the following questions:
- What other products serve a similar function to the product I’m planning to launch?
- How is my product going to be superior to those competing products?
Maybe your device will improve upon existing products with updated features that offer more value to the end user. Maybe you’re aiming to offer a cost advantage or a quality advantage.
To bring a medical device to market, it must have a competitive edge over other products aimed for the same purpose, and you will need to outline and plan to highlight that differentiating edge at the earliest stage - the marketability of your product depends on it.
Project Timeline and Costs
You need to have an idea of your project timeline before you begin. This should include estimates for each individual phase, not just your overall project timeline.
The amount of time you anticipate each individual phase to take matters because different phases have different capital requirements. Timing these phases correctly is key to managing your resources efficiently and creating an accurate budgetary estimate. Funding can also be dependent on achieving milestones, so you need to be efficient.
Your project budget needs to account for all expenses you might incur, and as we’ll expand upon below, it can be tricky to get this exactly right from the beginning.
However, with careful and thorough planning, you can start off with your best foot forward in terms of estimating all project costs at the beginning.
Items you may need to budget for include labor, materials, travel, training, consulting, product testing, regulatory compliance, clinical trials, and many more.
Taking the time to make a realistic budget is crucial to the success of your project, but new entrants in the industry don’t always envision all of the potential costs.
Having a general idea of the regulatory pathway is just as crucial. Yes, things may change as your product evolves, but the path to clearance or approval will have a significant impact on project timing and cost.
Testing is an example of something that can be overlooked in the beginning, and it is oftentimes much more expensive than companies realize. If your product is patient-contacting in any way, there is a very good chance you will need to conduct biocompatibility testing, and biocompatibility testing can cost tens of thousands of dollars, or even hundreds of thousands in some cases.
Do you plan to launch internationally? There are costs involved with registering a device in different countries, in the form of paying fees, obtaining a license, or simply paying someone to oversee the registration process. We will cover the device registration process in more detail later in this guide.
It’s common for new companies to miss their launch deadlines over budgetary hiccups. This is typically due to hidden quality management system (QMS) costs, such as repeat testing, spending weeks to compile Design History Files, etc. Overall, it is best to be proactive with a project plan knowing that it can change along the way.
Revenue Research
The other side of the coin for this process is calculating potential revenue. Calculating reimbursement and achieving commercialization are key components of your business model.
You’re not going to get the same market value for your product in one country that you may in another, and the same can even be true across certain U.S. states where prices may vary.
Planning for how much your product will cost in each individual market you plan to enter is an important part of your early-stage value proposition.
There’s a lot to consider with this part of your process.
Sometimes you’ll launch a great product, only to realize that hospitals in some of your target markets already have contracts with providers of similar products — the more information you can gather on these markets in various regions at this stage, the better.
AdvaMed has a great case study on Estimates of Medical Device Spending in the United States that I recommend you check out.
Create a Prototype
At this point, it’s time to get technical. You’ll want to consider prototyping your device to start troubleshooting problems and improve certain features.
There are many ways to get quick and inexpensive prototypes. Oftentimes, a device model will be drafted in CAD and then manufactured using cheaper materials than those which will be used for your end product.
An interesting trend we’re seeing in the industry recently is a huge shift toward 3D printing. A lot of companies get their prototypes printed using plastic materials, and in some cases even metal, which can be used to create a more realistic representation of the end product.
Once you’re satisfied with the basic design, it may be time to hire a contract manufacturer to build a more robust prototype. This can be costly, so make sure to budget for prototyping.
When you have a working prototype, you’re ready for basic feasibility studies. The purpose of these studies is to determine whether you should move forward with pursuing a full-blown project. Feasibility studies may influence your design choices at this time, and it’s not uncommon for people to introduce modifications at this stage.
One important thing to keep in mind is, if you’d like to leverage any of the results of these studies in a regulatory submission, you will want to make sure these are done under design controls, which will be discussed in great detail later in this guide.

Early Stage Considerations
As with any business, you will need capital and a team to bring your idea to reality.
There are many costs that people overlook early in the process. To successfully bring your product to market, you will need to consider costs associated with manufacturing equipment, testing, certifications with notified bodies, product registrations, clinical trial management, and many other items.
Now is the time to benefit from the creation of that aforementioned detailed business plan.
Secure Funding and Assemble Team
You would be surprised to know how many people out there are looking to invest in medical device companies. Capital can be obtained from many sources, including angel investors, venture capitalists, other companies, and grants. Having a solid, exhaustive plan will go a long way to relieve the concerns of potential investors.
Many new entrepreneurs feel as though they only have one shot to make this work. If that is the case, why not assemble an elite team?
Some decide to hire full-time employees, while others find consultants. There are many great consultants across all of the functional groups including quality, regulatory, design, manufacturing, etc. that have taken products to market, so it can help to leverage their experience.
In today’s world, it is easy to connect with people in the industry. This can be done online through LinkedIn or by attending an industry conference. Take advantage of any networking opportunities in your community and don’t be afraid to ask others for help. The medical device community is a tight-knit community and people are very willing to help each other out.
Another thing to remember is the Greenlight Guru team has a tremendous amount of experience with bringing products to market. In fact, our Guru team has been directly involved in the launch of almost 300 devices. Never hesitate to reach out if you have questions at any point throughout your journey.
Develop Regulatory Plan
At this point, you are probably energized and ready to attack the project with full force. However, reading the above heading probably turned that excitement to dread.
Regulatory strategy is one of the most, if not the single most, challenging aspects of launching a medical device. The good news is that many people have successfully navigated the regulatory waters in the past, and many more will navigate them in the future, including you.
You should have already done some research on the potential regulatory pathways for your device when you drafted your business plan. Now is the time to nail down a specific, detailed regulatory plan.
It is important to remember that every device is different and every device will have its own regulatory strategy. You are free to write your own story and do not need to follow a prescribed plan. Speaking from previous experience, the best strategies involve proactive planning with a clearly documented and sound rationale for the decisions that are ultimately made.
Selecting Markets of Interest
To no surprise, every major market for medical devices has its own specific regulatory framework. It can’t be that simple and straightforward, can it?
Your business plan should be the starting point for your regulatory strategy. Stick to the plan and don’t waste time trying to understand the regulations of markets that will not provide a good return on your investment.
Also, don’t feel like you need to launch in every target market on your list at the same time. Sometimes, a startup company can benefit from using one market as a stepping stone to enter another. For example, I worked for a company that had a Class IIb device in the EU, but a Class III device in the US. It only made sense for us to first start selling into the EU in order to help fund our IDE clinical trial in the US.
For any regulatory market where you plan to launch your medical device, it is important for you to understand, define, and document the intended use. A statement of intended use that clarifies exactly what the product is for is something that Greenlight Guru founder & VP of QA/RA Jon Speer discussed here in a podcast with guest, Mike Drues, Ph.D. The intended use is important for a couple of reasons:
- This helps to establish the scope of the device and will be useful when defining user needs, design inputs, and other design and development activities.
- The intended use is vital to supporting regulatory product classification, which ultimately dictates the pathway you will follow to get market clearance with FDA and other regulatory agencies.
To introduce a medical device to the market, there are a number of routes you can take depending on how your device is classified.
The classification depends on the perceived risk of your product type. Your classification is determined by regulators, and different regulators will, of course, have different stances on risk and different categories of classification.
The classification will inform you of the regulations you need to follow during design and development. This information will also give you more insight into how much it will cost to bring your device to market and the timeline in doing so.
As such, this has a major impact on your business model. We will break down the most popular markets now.
Determining US Classification
The Food and Drug Administration (FDA) oversees medical device regulations in the US - specifically, the FDA branch called the Center for Devices & Radiological Health (CDRH).
The CDRH assigns medical devices to one of three classes:
-
Class I (low to moderate risk): general controls
-
Class II (moderate to high risk): general controls and special controls
-
Class III (high risk): general controls and premarket approval (PMA)
The classification will determine the type of submission required for clearance, so it is very important to have a clear idea of what class your device will be early in the project. This will have a major impact on your project timeline and cost.
Most Class I devices are exempt from any type of premarket submission, however, there are a few that still do require a 510(k).
Most Class II devices, with a few exemptions, will require a 510(k) submission and clearance from the FDA before you can market your device. You will need to show substantial equivalence to another legally marketed device, often referred to as a “predicate device."
For Class III devices, a premarket approval application (PMA) will be required. You will likely need to complete a clinical trial, which the FDA will use to determine that the device is safe and effective.
There is one additional option that is gaining popularity amongst device makers. This pathway to market approval is for Class I and II devices in the US, known as the De Novo pathway. Eligible devices must be considered “novel” and thus not listed in the FDA classifications. To qualify, the device must also be considered low or moderate risk.
Take a look at our previous blog post on the De Novo pathway to learn more about whether this would be a suitable route for your device.
So how do you determine the classification of your specific device in the United States?
The FDA Product Classification Database is an excellent resource. In fact, it might be wise to bookmark that page.
You can search by device name, review panels, product codes, and much more. The output from that search will provide you with a wealth of information that will help you develop a regulatory strategy that makes sense for your product.

This process is not always as straightforward as it may initially appear. You will likely have a lot of questions or doubts, and that is okay. It happens to everyone.
Don’t hesitate to ask others in the industry who have been through it. Greenlight Guru’s team of medical device experts is happy to help remove the regulatory burden by guiding you through these types of difficult processes with confidence and ease.
Another thing often overlooked is the ability to solicit feedback early on from the FDA on your regulatory pathway through the FDA pre-submission program. This can be a very effective way to proactively work with the FDA to alleviate any concerns and be confident in your plan for getting to market.
Determining EU Classification
If you’re planning to sell into the EU, all requirements are outlined in the European Commission Regulation (EU) No. 2017/745, more commonly known as the Medical Device Regulation (MDR).
As many of you know, this is a new set of regulations and the transition period ends in May of 2020, so it is important to understand and implement these changes now.
There are four classes of medical devices outlined in the MDR:
- Class l (low/medium risk)
- Class lla (medium risk)
- Class llb (medium/high risk)
- Class lll (high risk)
In my opinion, determining your classification in the EU is much more straightforward than in the US. Annex VIII of the regulation contains classification rules that can be used like a flow chart to determine your classification.
Just like in the US, your classification will help you determine what is needed to get approval for your device.
To launch in Europe, you’ll need a seal of approval called a CE marking (or CE mark), which indicates the product complies with the essential requirements of all the relevant legislation outlined in the regulation.
Obtaining a CE Marking clears the product for sale within the single market. At the moment, the single market includes all EU states, as well as Iceland, Liechtenstein, Norway, and Switzerland.
For all devices, you will need to implement a Quality Management System (QMS) and maintain all project documentation in a Technical File. If you’d like to learn more about how to prepare your QMS for EU MDR, check out our Essential Guide to Preparing your QMS for EU MDR.
For any Class IIa, IIb, or III device, you will need to work with an outside party called a notified body. Notified bodies are accredited by the EU to conduct regulatory audits and ensure you are meeting the regulatory requirements.
The new EU MDR has impacted more than just device makers, making it seemingly more challenging for notified bodies to remain actively certified. The landscape is changing and there are fewer and fewer notified bodies available each day. You will want to plan ahead and find a notified body that can help you meet your project timeline.
Class I devices can be self-certified so your Technical File will not need to be audited by a notified body prior to obtaining a CE mark.
For devices in Class IIa, Class IIb, and Class III, you’ll need your Technical File or Design Dossier (for Class III devices) audited by your Notified Body.
Following a successful audit, which ensures you meet the regulations, you’ll be presented with a CE Certificate for your device.
Preparing for Other Markets
As aforementioned, every major market in the world has its own set of regulatory requirements. If you are planning to launch somewhere besides the US or EU, you will want to fully understand the requirements in those regions.
Some important things to consider when reviewing the requirements of various regions:
- Classification categories will likely vary from country to country.
- Some countries may require licensing or specific registrations.
- Clinical data might be expected in some regions.
- Physical representation through a local representative or agent may be necessary in certain countries.
- Obtaining clearance or approval in one region does not necessarily mean you will have an easier path to other regions.
QMS Implementation
By this point, you should know that you need to work under a Quality Management System (QMS) when bringing a product to market.
The FDA expects you to follow the regulations outlined in 21 CFR Part 820. Outside of the US, ISO 13485:2016 lays out all applicable guidelines.
The good news is there are many similarities between the two, and they are becoming more and more aligned over time. Being compliant to one usually sets you up for compliance to the other without a lot of extra work.
Now, let’s explore how to effectively implement a QMS to ensure you have the right structure and setup for full regulatory compliance to enhance product quality
QMS Structure
Many of you who have worked for multiple medical device companies have probably noticed something interesting… no two quality systems are alike.
A lot of people are surprised to find that you’re not required to use a “one size fits all” QMS. You have the autonomy to create one that works for your company and its needs.
The goal should not just be to follow the regulations but to ensure you are laying the foundation for a culture of true quality. A culture where the company’s employees naturally do the right things, and do them efficiently. The byproduct of that is almost always a safe and effective device.
So how can quality systems vary?
Since we already discussed creating a solid business plan, which includes the markets where you want to launch, a simple way to tailor your QMS is to focus only on the regulations that apply.
If you are only selling in the US, don’t even look at ISO 13485. If you know you want to sell in both the US and EU at some point, I recommend that you set up your QMS to comply with both Part 820 and ISO 13485.
It is also important to create a QMS that fits your company's size. A startup with four people doesn’t need a massive QMS right away - because they can all easily work in the same room, they can get away with a light QMS in a way that wouldn’t work for a company of 50 people, and so on.
Look at your short-term goals to determine what you will be focusing on first. Usually, the main focus of early-stage companies is to get their product through the regulatory submission process. In that case, there are some essential QMS processes that must be implemented from the onset:
- Document control and records management
- Design controls
- Risk management
- Supplier management
Start there and worry about the other processes (CAPA, Complaint Handling, etc.) when you are closer to manufacturing and product launch.
The last piece of advice I’ll mention is that when it comes to implementing a QMS, “keep it simple.” I have worked for companies where the SOPs have gotten so convoluted and complex that no one really knows what they should do. Some procedures may even contradict each other.
In fact, I have seen an auditor write an observation for a company that was following the regulations correctly, but its employees were not following their own internal procedures.
Overall, you need a set of procedures that will tell people what to do and how to do it. This helps ensure you are following the regulations and also ensures people are doing their work right the first time around.
How to Setup a QMS
In the past, most medical device companies used a traditional “paper” QMS to bring devices to market because that was the only option available. A paper QMS does not necessarily consist of just binders of paperwork but can refer to systems stored on servers, DropBox, Google Drive, etc.
If you’ve already used a paper QMS, you know that it can be very cumbersome and inefficient.
In addition to signing, scanning, and storing relevant documents, this method necessitates the need to constantly reference and update procedures throughout your company’s lifecycle.
There’s a lot of back-and-forth between different team members, and when done manually, this can be a significant pain point. I’ve even seen people retype entire procedures because they could not find the original source file.
With larger teams, it’s not uncommon for updates to be done simultaneously — and the last thing you want in an audit or inspection is to discover that your engineers are using different versions of the design and development procedure.
With a purpose-built digital QMS solution like Greenlight Guru, everything is updated in real-time, eliminating the risk of users working on multiple versions of the same procedures. All of your procedures are easy to find and with a simple click of a button, you can reference any record through the search function.
In addition to the software, Greenlight Guru can also provide pre-written SOP templates, all of which have been audit tested and used by dozens of companies in hundreds of markets across the globe.
![UG-BMDGM_product development phase_3]() Product Development Phase
Product Development Phase
 Product Development Phase
Product Development PhaseNow that you have a proper foundation through an established business plan and focused quality management system, it’s now time for the fun part… taking your idea and making it a reality.
The product development phase can be daunting though, as it consists of many factors. The question everyone always asks is “How long is it going to take me to get this product launched?”
In my opinion, that is not the question you should be asking yourself. Instead, you should ask, “How do I best set up milestones to make sure I deliver a quality product to market?”
Developing a medical device is not something you should rush in order to meet some arbitrary project deadline. You need to make sure you have a good plan in place and take your time. Doing it right the first time can save a lot of headaches later in the process.
Most people get one shot to bring a medical device to market. If you launch your product with any type of issue, the chances for a do-over are unlikely, especially in today’s world where information travels so quickly.
When it comes to product development, it is best to get organized early and be proactive about what you’ll need to do. This will form good habits for your entire team and make for a smooth development process.
Design Controls
As I talk to more and more people in the industry, it seems like design controls get an unfair bad rap. I cannot tell you how many engineers have told me they should have entered a less regulated industry. “I didn’t go to school to do paperwork” is something I heard quite a bit when I first entered the workforce.
Many people think this way because they wait to compile all design control information until the end of a project. For them, it truly is a worthless exercise with no added value. Half of the information is incorrect or doesn’t exist at all. Why do you think there are so many remediation projects taking place these days?
Those that “get it” start documenting design controls early in the project and use them to develop a better product. No matter what method of design you are using, I guarantee you are thinking of all aspects of design controls in your head, so you might as well document them early.
The FDA and ISO both have similar statutes when it comes to design controls, as shown in the graphic below.

Both emphasize the need to have full traceability of design controls. Your project will change and grow as you navigate your path to market, and you need to keep track of the changes you’re making to your design along the way.
Most people use some form of a design control traceability matrix to ensure everything is documented and traceable. I like to think of this matrix as the “executive summary” of your device design. This is an important artifact because it is often one of the first things an auditor or inspector will look at to understand your device.
If you need a more in-depth overview of the design control process, I highly recommend reading our Ultimate Guide to Design Controls, which explains and walks you through every step of the process.
Many people use Excel to build their matrix, which seems crazy in mind since Excel was built for crunching numbers. It can be burdensome to create and maintain, especially when many stakeholders are involved in updating the information.
Greenlight Guru has a purpose-built traceability matrix (seen in the image below) that allows you to easily view and demonstrate the relationship between design control elements, such as user needs, design inputs and outputs, and all verification and validation activities.

Click here for a free personalized demo of Greenlight Guru's Multi-Level Design Control Software
Next, we will break down the different aspects of design controls.
Design and Development Planning
If you haven’t figured it out by now, planning is important for every aspect involved in bringing a device to market. It should be no surprise that you need to plan for documenting and reviewing your design controls.
Much of the planning will occur early in the project, but it is important to know that this plan should not be static. The plan should be reviewed and updated as your design and development work evolves.
A good plan will include a description and timeline of all development activities, information about who is responsible for the different activities, and clear expectations for when you will hold design reviews.
User Needs
You may have noticed that user needs are not actually called out in any of the regulations. Because of that, they are sometimes overlooked and people jump right into device specifications.
It is very important that you first ask yourself “why” you are making this device. You need to understand the needs of anyone who may interface with this device, clinicians, patients, lab personnel, etc.
Understanding the “why” will help you identify the intended use (purpose of your device) and indications for use (medical conditions that your device is aimed to treat, prevent, diagnose, cure, or mitigate).
Greenlight Guru founder & VP QA/RA, Jon Speer outlines 7 key questions below that you should consider when defining user needs. As you think of the answers to the question, you want to also consider that all user needs will need to be validated prior to launching your product.
- What do you want the device to do? Does it cure a disease completely or help with the symptoms?
- Who is going to use it? Are the users young or old? Do they have other medical conditions in addition to the one you’re treating for? These questions will make slight differences to the design and function of any device.
- When will it be used? Occasionally or all the time?
- What important attributes or features should be considered? This is the nitty-gritty, really making sure you think through all the ways this device could help people.
- How will the user interact with the device? Will the user be able to work it on her own, or will she need professional help with it?
- What type of procedures will the device be used for? Is it for surgeries, rehab, or daily life, and for what types of activities in these categories?
- Is the device used once, or repeatedly? This will make a difference in manufacturing and quality requirements.
Design Inputs
Design inputs are near and dear to any engineer. They are the foundation of your device design and how you document all of the product requirements for the device.
Coming up with all of the design inputs for your device is no easy task and I suggest checking out this guide on design inputs and outputs to help you plan and define them.
Everyone feels as though they understand design inputs, but many people still struggle to come up with a good definition. According to the FDA, “Design inputs mean the physical and performance characteristics of a device that are used as a bases for device design.”
Good design inputs are clear and objective, and they’re written in such a way that you can easily prove or disprove them, just like a scientific hypothesis.
There is no specific format you need to follow when drafting them, but it’s generally useful to use language like:
“The device will have an outer diameter of 0.8 mm”
“The device must trigger an audible alert if the battery drops below 20%”
There are many resources you and your team can use to develop your design inputs, such as:
- Industry standards
- Regulations
- Previous projects/products
- Competitor products
- End users
- Prototypes
These inputs will be put to the test when it comes to design verification, so you need to document your inputs in such a way that you can prove they’ve been met when you reach the verification stage.
Commonly considered design inputs include the size and shape of the device, material characteristics, and anything related to how the device functions. However, there are some other aspects of the design you also want to consider.
One important consideration is labeling, which few people realize is considered to be part of the device design.
Labeling doesn’t just refer to the label on your device but also includes instructions for use, marketing materials, graphic material accompanying the device, and any other information used to describe your device to the end-user or regulators.
When it comes to labeling, you will want to familiarize yourself with all of the FDA’s requirements for a Unique Device Identification (UDI) System. Additionally, the EU MDR requires that all devices sold in Europe be issued a unique identification number to help with exchange of information including product registration, declaration of conformity, economic operators, vigilance reports, and post-market surveillance.

Medical device packaging is another aspect of the design that can sometimes be overlooked. As with everything else, leaving the planning of this process until the end can result in delays and needless expenditures. As soon as your device shape and size have been designed, you will need to start planning your packaging inputs.
Related to packaging, if your device is provided sterile you will want to make sure you think about all of the sterilization requirements. The go-to standards for packaging sterilized devices sold in Europe and the US are ISO 11607-1 and ISO 11607-2.
Design Outputs
Design outputs are the drawings, specifications, or manufacturing instructions associated with your device. Imagine you’re already working with an external manufacturer to physically produce your device - outputs are what that manufacturer will use to do that, and the outputs must describe all of the product's assemblies and sub-assemblies.
Another way to think of design outputs is that they are the “recipe'' for making your device. Your outputs will go on to form the basis of the device master record (DMR) when you reach the production phase.
So many companies work in silos where the engineers develop the specifications without any feedback from the manufacturing team. I remember being told many times early on in my career that engineers are great at coming up with fancy designs, but they have zero clue how to actually make anything.
Be that as it may, make sure you involve the manufacturing team, whether it is an internal team or an outside vendor, early in the process so they can provide feedback on the design outputs.
The ultimate goal should be to hand that DMR, or packet of design outputs, over to your manufacturing team so they can make the device consistently to specification and in an efficient manner. This can save your company vast amounts of time and money.
Design Verification
Arriving at this stage is a huge accomplishment.
However, for many, design verification can be frustrating, time-consuming, and flat out confusing. It’s generally the part of your journey that will consume the most time and resources. It is also a common point where things can go wrong and delay the project.
At this stage, you’ll be drafting test protocols, running tests, verifying results, and writing reports. Some testing will take place internally, while other testing will be outsourced. Again, a good verification plan is vital to stay organized and on track.
Ideally, you should have started drafting your verification plan when you created the initial project plan — waiting until just before verification is too late and one of a few common pitfalls that can create major pain points in what is already a challenging process.
The main purpose of the verification process is to confirm your design outputs meet your design inputs.
While inspections, scientific analyses, and various other activities can make up your verification items, much of this stage is going to consist of product testing.
You will do a lot of testing that is specific to your device. These may include aging testing, material strength testing, fatigue testing, and many others.
There are also many tests you will need to consider, regardless of the type of device you are developing.
Biocompatibility is something that needs to be addressed for every single product that hits the market, regardless of classification. You need to prove that all materials in your device are biocompatible, even if those materials have been tested and approved for use in other medical devices already on the market.
People often believe that they don’t need to test because their product is similar to an existing product or uses the same material, but that is not the case unless you can prove that your manufacturing process is the exact same as that of the other products.
If your device uses electricity, it falls under medical electrical equipment (MEE) and you’ll need to demonstrate that it meets user needs and intended uses in that regard.
MEE sold in the US and EU must comply with safety standard IEC 60601-1, or a subsidiary of that standard. Greenlight Guru has a 15-step checklist to help you gain IEC 60601-1 approval.
Design Validation
It is very easy to confuse design verification and design validation. The important distinction is while design verification focuses on your design inputs, design validation is done to prove your device conforms to your defined user needs and intended uses.
This is usually done near the end of the project. In fact, the regulations specify that design validation should include testing of production units, or their equivalents, under actual or simulated use conditions.
Validation activities can be hard to define, so one thing I always recommend is looking up summaries of the regulatory approval applications for similar products. You can often find brief summaries of the type of testing those products underwent prior to being released to the market. The FDA’s 510(k) database is an excellent resource for this.
Don’t be afraid to get in touch with regulators or your notified body either. While regulatory bodies can’t directly advise you on your process as consultants, you can seek feedback on your proposed testing before going ahead with it.
The more research you can do with regard to testing prior to this phase, the better.
Human factors is frequently talked about and something you want to consider during the validation phase. The FDA stresses the importance of enhancing patient safety by introducing human factors engineering processes in your design controls and risk management.
This means that you must ensure that your device can be used safely and effectively by people who are representative of the end users under expected use conditions.
This is done by conducting studies and tests, often using mock-ups, early prototypes, or partial designs of your device with study participants who represent your actual end user.
Can your study participants easily understand and follow the instructions on the labeling?
Is any training required to use your device?
These are the kinds of questions answered at this stage of the validation process.
Sometimes simulated use with mock-up designs isn’t enough to pass validation. Clinical trials (or clinical investigations) are not always necessary but can be used as validation activities.
In the US, around 10 - 15% of Class II devices undergo clinical trials, while all Class III devices rely on clinical trial data to get to market. It is important to note that all Class III devices in the US must secure an initial device exemption (IDE) from the FDA before beginning clinical trials, unless exempt from IDE per 21 CFR 812.2(c).
Design Reviews
Regulatory standards dictate that you must implement design reviews, and doing so allows you to monitor the progress of your medical device project throughout its development.
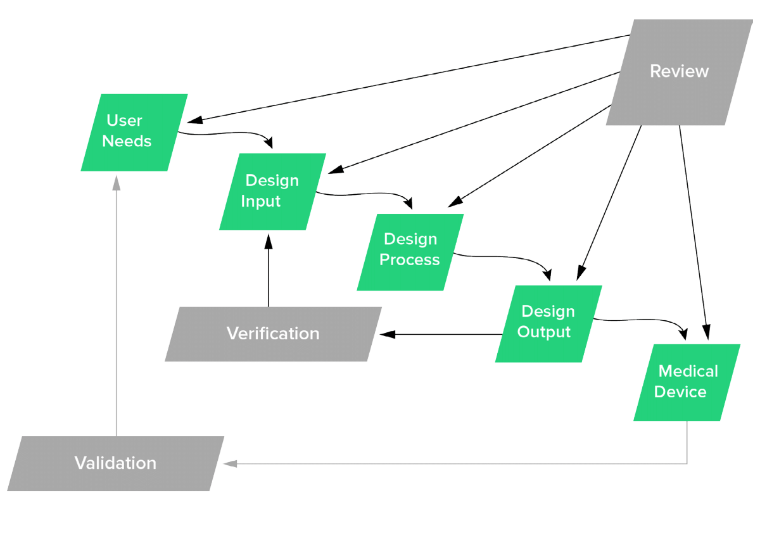
Waterfall Diagram in FDA's Design Control Guidance For Medical Device Manufacturers
There is no fixed answer to the question of how many design reviews you need. The above FDA waterfall diagram indicates a good guideline of when a design review would be ideal, which includes:
- After User Needs & Design Input Requirements are created
- After Design Outputs are defined
- Prior to initiating Design Verification
- After Design Verification and prior to Design Validation
- When ready for Design Transfer.
Each project is different, and these may not be practical depending on the agility of your project, but a good rule of thumb is to conduct a review at each of these milestones. You can think of each milestone as a checkpoint to ensure that your device is safe and effective.
FDA 21 CFR Part 820 and ISO 13485 have similar guidelines on design reviews. A key difference is that the FDA specifies that you must appoint an “independent reviewer” to attend your design reviews.
From an internal standpoint, multiple departments are involved in design reviews.
- Design: Your design team will review design controls, communicate ideas and concerns to other departments, and solicit feedback on the current design.
- Quality: The quality team has to be represented in the design review process to rigorously enforce quality standards.
- Regulatory: Regulatory staff will ensure that everything is compliant with the relevant regulatory standards.
- Manufacturing: Changes made during the design reviews will inform the manufacturing department of the way the device will be made and ensure that it’s actually possible to manufacture the design outputs, which define the medical device’s components and how it will be assembled, and to help evaluate the cost of doing so.
- Marketing: Marketing staff will also be present, as any changes made to the product development during reviews may influence their approach to labeling and packaging which must comply with all standards.
I’ll be honest - design reviews can be a project pain point, often carried out in live meetings with a lot of staff.
You can handle this process solely through live meetings with a paper QMS, although, again, using a digital QMS allows staff members to work on separate components at the appropriate time..
You should aim to keep design reviews to 60 - 90 minutes. Sharing any relevant documentation with design review attendees a week in advance will help you achieve this by preparing everyone ahead of time.
Stick to a fixed agenda and use a design review checklist to address each point methodically as you go.
Design Changes
You are expected to identify, document, and approve any changes to the design throughout the process. This applies to both pre- and post-launch of your product.
Part of the approval process should include an assessment of whether any verification or validation activities need to occur or be repeated. Overall, you need to make sure that all devices used in the field are no different than the device that was verified and validated.
Design History File
Everything you have done so far as part of the design control process should be documented and organized into design history file (DHF). The DHF demonstrates that the device was developed in accordance with both the design plan and the requirements of this part.
Many companies spend the last couple of weeks of a project compiling all of the documentation and stuffing it into multiple binders. They then shove those binders in a closet, or maybe even ship them to an offsite storage facility, and have zero plans to open them again… unless, of course, an auditor shows up.
That is all wrong for multiple reasons.
A DHF should be a tool and resource for the team throughout the entire project - think of it as the source of truth for your medical device. If you’ve designed your DHF in an accessible way, your team can easily refer to it when needed to help answer queries and aid development.
This also has utility after the product launch. Yes, even the best medical devices are likely to have some sort of design change for one reason or another. In fact, most regulators expect your DHF to be a “living file” that can be accessed and updated at any time. That certainly doesn’t sound like a dusty binder at an offsite storage facility.
Overall, you need to make sure your DHF is accurate, organized, and easily accessible.
Greenlight Guru allows you to custom-build your DHF and work on it in real-time. It does this by automatically compiling the DHF as you go, meaning that by the time you reach the end of your project plan, the DHF is already built out.
You will also have a true “living” DHF with full traceability between your design control items and the individual documents, like drawings, test reports, etc. that relate to those items.
Using an electronic system design specifically for design controls feels much less like “paperwork” that engineers dread, and allows you to focus on the important stuff, like designing the best product possible.
Risk Management
Simply stating the words “Risk Management” has garnered more sighs and groans than I can even count. Everyone in the industry has a different interpretation of what risk management actually looks like.
The only thing almost everyone can agree on when it comes to risk management is that no one likes to do it. Much of that disdain is due to the processes or methods people use.
We can all agree that patient safety should be at the center of everything we do. Most engineers are likely already thinking about mitigating risks when they start picturing the device in their heads.
Creating devices that save lives and improve the quality of life for end users is a very rewarding task, and it comes with a lot of responsibility. Your design decisions should always consider any risk implications, which I’ll expand upon below.
What is Risk Management?
As we approach risk management, it is important to first understand the basic terminology.
- Risk is the combination of the probability of occurrence of harm and the severity of that harm
- Hazard refers to a potential source of harm
- Hazardous situation refers to circumstances in which people, property, or the environment are exposed to one or more hazard(s)
- Harm is physical injury or damage to the health of people, or damage to property or the environment
- Severity is a measure of the possible consequences of a hazard
These definitions are all outlined in ISO 14971, the international standard on risk management. Most of the companies I work with are more familiar with the analysis method called failure mode and effect analysis (FMEA) than they are with ISO 14971.
Some have heard of both FMEAs and ISO 14971 and believe that they are much the same, but this is not the case. Unfortunately, during my career as a product developer, I once had to stutter my way through trying to explain to an auditor why we included “detectability” on our risk matrix. You want to make sure you’re using the correct ISO 14971 terminology in your risk documentation to ensure that regulators can easily understand your procedures.
Now that we’re familiar with these key terms, let’s explore how to plan for and mitigate risk.
Risk Management Planning
Regulators in nearly every major market expect you to have a well-defined risk management process and risk management documentation to support it.
A risk management plan involves the following:
- Definition of the product or products you’re planning for
- Description of product’s intended use
- Description of all risk management activities
- Roles and responsibilities (identify your risk management team)
- Criteria for product’s risk acceptability
- Methods to verify risk control
- Definition of how post-production information will be recorded and used
Risk Management File
A risk management file (RMF) is the section of your QMS where you keep all your risk management activities, records, and other documentation.
Your risk management plan, risk analysis, risk evaluation, risk controls, evaluation of overall risk acceptability, risk management report, and production and post-production risks will all live here.
It is also crucial that you consider the overall impact on risk when making all design decisions. Because of this, you need to make sure there is a clear connection between your design controls and risk management information.
Greenlight Guru is purpose-built to comply with ISO 14971 and accommodate the creation of a comprehensive risk management file that is fully integrated with design controls. Greenlight Guru is the only QMS software that fully aligns with ISO 14971. You can learn more by requesting a free personalized demo of our Risk Management Software.
Risk Assessment
At Greenlight Guru, we view risk assessment as being comprised of two parts: risk analysis and risk evaluation. These aspects of risk assessment can be managed in tandem with one another, making for an effective, efficient assessment process.
The all-important task of identifying risks in medical device development starts with risk analysis.
ISO 14971 Annex G identifies several ways to perform risk analysis, including preliminary hazards analysis, fault tree analysis, and FMEA, which I mentioned earlier. As you can see, FMEA is one small part of the overall risk assessment and should not be the only thing you do.
The first step in completing a proper risk analysis is documenting an intended use statement which describes what your device is to be used for.
Once you’ve crafted your intended use statement, you’re likely to have a better idea of how people may misuse your device, accidentally or otherwise. You should consider hazards arising from your product, being used as intended, as well as hazards from potential misuse.
Consider each step that an end-user will take when using your product, and identify any hazards that may arise at each point, either endangering the end user, the environment, or damaging property.
You can find a list of example hazards in ISO 14971 Annex E.
With your hazards identified, consider situations where these hazards might crop up. Here’s an example of a hazard and a hazardous situation taken from ISO 14971:

At this point, you will need to identify the harms that arise from hazardous situations. Remember that harms not only include physical injury or damage to the health of people but also damage to property or the environment.
After the harms are identified, you need to estimate the risk for each harm. In estimating risk, you’re considering the severity of each risk, and the probability of that risk occurring.
Here’s a table showing an example of the different levels of severity:

The table below shows one way you can measure the probability of occurrence:
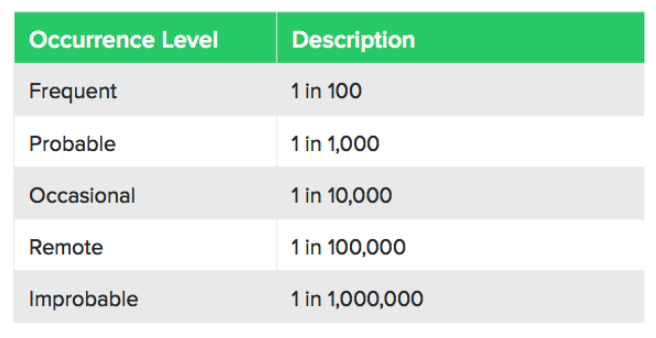
It can be tricky to measure the probability of occurrence, but you can take inspiration from similar products, regulatory data, industry standards, scientific white papers, end-user expertise, and supporting test data.
We go into more detail on this in our Definitive Guide to Risk Management.
Even if you identify a risk, it’s not always possible to completely mitigate it, and there is a certain amount of acceptable risk inherent in medical device development. Risk evaluation is how we determine that.
Below is an example risk acceptability matrix used to categorize risks:
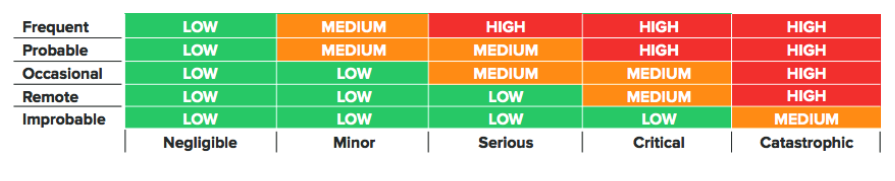
Risk controls are measures that you take to reduce the risk of your device to a minimum level, increasing the safety of your device.
You should prioritize your risk controls as follows:
- Inherent safety by design
- Protective measures in the device or manufacturing process
- Safety information in labeling or instructions
First and foremost, the best way to reduce risk is through safe design, with labeling to warn against risks being the last resort. Generally speaking, you can and should include multiple risk controls to reduce a single risk.
Build Supplier and Partner Network
Companies of all sizes rely on partners to bring their medical device to market. These partners can be contract manufacturers, distributors, test labs, consultants, and many more.
Partners can play a crucial role in your business, and that can be a bit scary - after all, your company name is going to be on the label and you are going to have to answer to both regulators and customers.
Even though you have little control over how an outside party does business, you can still take action to ensure that you’re working with the right people and that they’re on the same page as you when it comes to your product development.
The first step is implementing a robust supplier management process. Just like with product design, you need to take a risk-based approach to choosing and managing suppliers.
One thing I recommend when identifying suppliers is to make sure you do your research and shop around. If one supplier is significantly cheaper than the other, is that a business opportunity, or a red flag?
Choosing the wrong supplier can wreak havoc on your product development, so take your time and choose wisely. Look up reviews, talk to people they’re in business with, and perform general due diligence before making a decision.
You want someone on the approved supplier list with a quick response time, good value (in terms of cost, lead time, quality, or all three), and a good reputation.
It is also smart to complete regular audits on your suppliers. Your quality department should include suppliers and economic operators when it comes to quality control checks. Consider the quality of the product, the timeliness of production and development, and the conditions in which it’s produced - leave no stone unturned.
Regulators expect you to be able to prove that your products were made in a way that complies with regulatory standards, so it’s important to be thorough here.
When it comes to identifying consultants and distributor networks, it is again wise to use your connections. Find someone who has experience in your field. When it comes to navigating murky regulatory waters, there is nothing more valuable than having a resource who has successfully brought a product to market.
Develop Relationships with End Users
A common mistake that a lot of companies make is they develop their product in a vacuum. They think they know what the user wants and maybe have some initial conversations with clinicians and patients, but the communication stops there.
Developing relationships with end users is important for a couple of reasons.
One, you want to make sure you use their feedback to improve your design throughout your development project. As we discussed before, they will play an important role in your design validation.
It might even be best to set up an official design team with clinicians. Not only will they help with product improvements, but they can also be a voice for your company by speaking at conferences, talking about your product with their colleagues, and even publishing journal articles based on your product.
Another important reason to build relationships with end users early is so they can be advocates of your product. Patients, for example, do much more research now than in the past. They are not just relying on their doctor to tell them what they should get. Social media has been a powerful tool to bring information directly to patients and can be a place to get positive reviews on your product.
Regulatory Submission
There is no feeling quite like submitting your packet of information to the regulatory body. It is a mix of relief, excitement, and extreme anxiety. When I was working on my own products, I remember checking my email hundreds of times a day hoping for a response. It is like waiting on your final exam grade in college but with much greater ramifications.
If you correctly set up your QMS and were proactive about documenting design controls and risk along the way, the submission itself is not that bad. This is another area where there is no one size fits all. You need to tell the story of your product in a way that is easy to understand for the reviewer.
US FDA
As we discussed previously, your device classification will dictate the type of submission to the FDA.
- Class I: These devices are simple, with minimal risk to the user. They are subject to the general regulatory controls of medical devices and typically do not require any premarket submissions.
- Class II: Devices in this class pose a moderate level of risk to the user, and all of them require a premarket notification (510(k) submission) before they can be legally marketed. Items like pregnancy testing kits, intravenous kits, sutures, and powered wheelchairs could fall into this category. These devices are important for health care, but a malfunction would be unlikely to cause critical harm to a patient.
- Class III: Class three devices are typically either implanted medical devices or those that sustain life, like an implantable pacemaker, blood vessel stents, or other implanted device. Devices in this class are seen as the highest risk for patients, as any problems with the device could lead to significant adverse outcomes for the patients. Class III devices require a PMA submission before being marketed in the USA.
The purpose of a 510(k) submission is to provide the FDA with documented evidence which proves that your medical device is substantially equivalent to a predicate device, one that has already been approved for marketing by the FDA.
Proving substantial equivalency means that you'll need to compare and contrast your device with the predicate device, and while laboratory testing is a typical requirement, human testing is usually not needed for 510(k) submissions.
Information from your documented Design Controls process, such as intended use, indications for use, design inputs (learn how to define them here), and design verification are all useful inputs for your 510(k) submission.
The FDA processes 510(k) applications in 30-90 days. In some cases, there can be an extended period of back and forth discussions, delaying the time to actual clearance. This is another thing to consider when you build your project timeline.
A PMA is more in-depth than a 510(k) - it is used to prove that a new device is safe and effective for the end user and typically requires clinical trials with human participants along with laboratory testing.
The standards here are much higher than for 510(k) submissions, and the FDA has just 180 days to accept or reject the application.
Another less commonly used option, the De Novo pathway, is a classification process that uses a risk-based methodology for the approval of new and novel devices to be sold in the market.
Generally speaking, companies using the De Novo pathway do not qualify for 510(k) clearance, due to the fact there is not an existing predicate device substantially equivalent already on the market. Since they are not showing equivalence to a predicate, companies must be prepared to argue their robust risk mitigation strategy when submitting a De Novo request.
EU CE Marking
The route to CE marking is much more straightforward in my mind. As part of your regulatory plan, you have already determined which EU directives apply to your device and determined the classification of your device.
Implementing a quality management system that is compliant with ISO 13485:2016 is a crucial piece that should already be done at this point. You will need to identify a notified body and have them audit you to ISO 13485 so you can receive your certification.
Class I devices can be self-certified so technical file documentation will not need to be reviewed by your notified body. However, the technical files for Class IIa and IIb devices or design dossiers for Class III devices will need to be reviewed by your notified body to obtain a CE certificate.
The technical file/design dossier is very similar to the DHF, but there are some differences. One is your notified body often expects a specific format for the documentation. Despite that, I often took information directly from the DHF and rearranged the format to please my notified body.
One important step that is sometimes overlooked is you need to establish a physical location in Europe or appoint a European representative for your company.
You then need to prepare a Declaration of Conformity (DoC), stating that your device complies with the appropriate directives.
At this point, you can contact your notified body to schedule the technical file audit. Assuming all goes well, they will submit a CE certificate for your product that is valid for three years.
![UG-BMDGM_product launch_4]() Product Launch
Product Launch
 Product Launch
Product LaunchSo much work goes into designing a product and submitting a regulatory submission that many people assume the work stops there. However, there is still much work that lies ahead to make sure there is an adequate supply of your product and that it is actually used in the field. In order to execute a successful product launch, it is again wise to adhere to a plan.
Design Transfer
One of the main goals of any company is to sell their product… a lot of it. If you want to sell in high volumes, you need to ensure you have an efficient and reliable manufacturing process running behind the scenes.
The design transfer process is all about handing over the necessary information to your manufacturing team so they can make your product. It is vital that the products made during the manufacturing stage match the products made during the verification and validation phase.
We all know that design transfer is a regulatory requirement. Both 21 CFR 820.30 and ISO 13485 describe design transfer as a way to ensure design outputs are transferred into suitable production specifications. It also just makes good business sense.
As we discussed in the design outputs section, you want to include your manufacturing team early in the process. They are the best resources to tell you whether a design makes sense from a manufacturing process.
They can help you find the best materials, offer insight into geometries that will help facilitate a smooth manufacturing process, and can offer just general design for manufacturing suggestions.
The main goal of the design transfer activity is to finalize your device master record (DMR), defined by the FDA in 21 CFR Part 820.3 (j) as “a compilation of records containing the procedures and specifications for a finished device.”
If you follow the instructions in this guide and begin building design transfer early on in your design review process, the actual design transfer meeting can simply be a stamp of approval on your design outputs.
Setup Distribution Network
The phrase “your product won’t sell itself” is one you’ve probably heard before. Although the idealists in this world might disagree, it is a good mindset to have when launching a product.
I haven’t met a lot of engineers who are too fond of selling, and I suspect many of you feel the same way. It would be great if you could launch your product and move on to designing the next life-changing technology.
If this is how you think, you are going to need a solid network of sales and marketing professionals. With the ever-changing global economy, there are many companies throughout the world that have a proven infrastructure in place to focus on selling products and growing revenue.
Not only can distributors help with the actual selling process, but they can also often provide marketing support, and help organize other events such as exhibiting at conferences and running product training courses.
This is yet another instance where networking and using your connections is so important. Depending on where you’d like to sell your product, do everything can to research and connect with distribution companies in those areas.
Develop Marketing Materials
Again, your product won’t sell itself, especially if no one knows about it.
You already know about so many of the complexities involved with the medical device industry. There is certainly no exception when it comes to the marketing and selling process.
The buyers of your device may be hospital systems, physicians, patients, distributors, and maybe even the government in some countries. Because of this, you need to have a marketing plan that considers all of these stakeholders.
You will need everything from social media posts to catch the attention of patients to technical documentation so a physician can better understand the capabilities of your product.
Remember that marketing materials may also be reviewed by regulatory bodies. Even your website may be reviewed so it is important to not make any claims that are not backed up in your submission.
Product Registration
Obtaining clearance on a 510(k) and receiving a CE certificate does not necessarily give you the green light to start selling your product. You need to make sure you register your device and/or facility in the different countries you’d like to sell.
The FDA requires companies involved in the production and distribution of medical devices to register annually with the FDA. Additionally, many countries throughout the world require registration or licensing to sell your product within their borders.
The distributor you select will be a great resource for learning more about registration. This valuable resource can often can even provide services to register the device for you.
Reimbursement Strategy
Reimbursement is one of the most confusing topics when it comes to launching a device. You obviously need to make sure you are going to get paid for your device and it is important to think of this early.
One thing that you might find surprising is you will likely sell your device for many different prices. For example, a knee implant might sell for $2000 to a hospital in New York City and $1000 dollars to a hospital in Indianapolis. That very same knee implant might sell for $300 dollars in India and the government in France might tell you they will buy it for $500.
There are so many factors that go into reimbursement. It is important to understand the process in the different regions you’d like to sell. Most countries have a coding system that will dictate the costs for procedures and products.
You also want to make sure you integrate your reimbursement strategy with your regulatory strategy. A lot of times the reimbursement codes are tied to the intended use of the product.
Reimbursement is not a fun topic and it is important to connect with experts to prevent any headaches when it comes to launching your product.
![UG-BMDGM_post launch considerations_5]() Post Launch Considerations
Post Launch Considerations
 Post Launch Considerations
Post Launch ConsiderationsOnce your product is being sold and used, you have a responsibility to oversee its efficacy. While the stringent regulatory process is as thorough as possible, the fact is that clinical trials carried out in the validation phase involve relatively few people, or none at all in the case of simulated use trials.
It’s not possible to fully recreate market performance conditions in the trials, and that’s where postmarket surveillance comes in.
Postmarket Surveillance
Postmarket surveillance (PMS) refers to the responsibility of device manufacturers to monitor the performance of their own product once its reached the market. Medical device companies are often obligated by regulators to do this and with good reason.
When it comes to product surveillance in the US, the FDA has established MedWatch, a web-based portal designed to facilitate PMS. Medical device developers can submit reports of any reported risks, failures, or nonconformance incidents that may occur during the use of the device. The FDA also carries out independent PMS.
The FDA requires device manufacturers to carry out PMS for Class II or Class III devices that fit the following criteria:
- Devices that could cause serious health consequences upon failure
- Devices that are used in pediatrics
- Devices that will be implanted in end-users for over a year
- Devices that are intended for life-sustaining or life-facilitating use outside of medical facilities
Class I devices are typically exempt from postmarket surveillance requirements.
In Europe, the process is referred to as post-market clinical follow-up (PMCF), and unlike the US, it’s obligatory for all medical device manufacturers in that market. Manufacturers must either provide evidence of PMCF or a justification as to why it was not done.
Technically, PMCF applies to devices that fit the following criteria:
- The device carries novel medical technology
- The device carries a high product-related risk
- The device is used in high-risk populations (children or the elderly)
- The device is being used by a more varied demographic than that used in the clinical trial
In practice, these criteria apply to virtually every device on the market. The EU MDR, which was enacted in 2017, heavily focuses on PCMF in the European Economic Area, and device manufacturers operating in Europe need to be compliant.
The EU MDR obligates device manufacturers to operate using a QMS and have a robust post-market surveillance program. Greenlight Guru specializes in facilitating EU MDR compliance by securing documents in accordance with industry standards to ensure they’re accessible only with proper authorization, which is also beneficial in speeding up the auditing process with notified bodies.
Manage Quality Events
When you have fully ramped up production and are selling your product in multiple markets, you will need to track and trend a variety of quality events, including nonconformances, customer feedback/complaints, and CAPAs.
This is the point where many companies expand their QMS and add procedures for these areas. As with the product development phase, clear documentation is vital.
Nonconformances
A nonconformance is an incident wherein your product has failed to meet specifications.
Having robust internal operations procedures and a solid supplier management process and can offset nonconformances, but the truth is, they’re likely to occur at some stage.
First off, you’ll need a way to identify nonconformances. Random sampling, batch testing, and internal audits are all some of the industry best practices. Nonconforming products then need to be evaluated to determine whether an investigation is required.
You should create a procedure that outlines roles and defines responsibilities in the event of nonconformance. Someone needs to be in charge of reviewing products and disposing of any that don’t meet requirements, and this SOP needs to be included in your QMS.
Beyond disposing of faulty products, you need to be able to rewrite whatever procedures or work instructions that may have led to the production of a nonconforming product. You will need to identify the root cause of the issue and update the processes to reduce or eliminate similar nonconformances in the future.
Customer Feedback
A key aspect of PMS management is soliciting customer feedback. Most companies are good at responding to customer complaints, but not many are as proactive as they should in reaching out for general feedback. It is good to set up a plan for gathering feedback proactively because it can help identify problems before they become systemic.
Of course, there are other benefits to soliciting feedback. Sometimes, customers may have helpful suggestions for aspects of your device that, while posing no risk, could simply be improved upon.
Design suggestions such as ergonomics, color, and material can help inform your product development long-term. Remember, the end-user has a unique perspective on your device that not even you or your engineers will fully grasp - so pay attention to them!
There are many ways to solicit feedback including carrying out surveys of your end-users, as well as employing focus groups.
Complaint Management
If you’re launching a product, it’s inevitable that you’ll receive a customer complaint. Despite your best efforts, cautious planning and design procedures, it’s bound to happen and that is okay.
While you can’t prevent complaints entirely, you can handle them in the best way possible both from a regulatory and business standpoint.
Complaint management regulations are outlined in FDA 21 CFR 820.198 and ISO 13485:2016 section 8.2.2, and both regulations stipulate that you need to maintain a customer complaint file as part of your PMS.
This file needs to be well-maintained, as regulators are absolutely going to want to view how you handle your complaints.
Here’s a flowchart depicting an overview of the complaint management process:
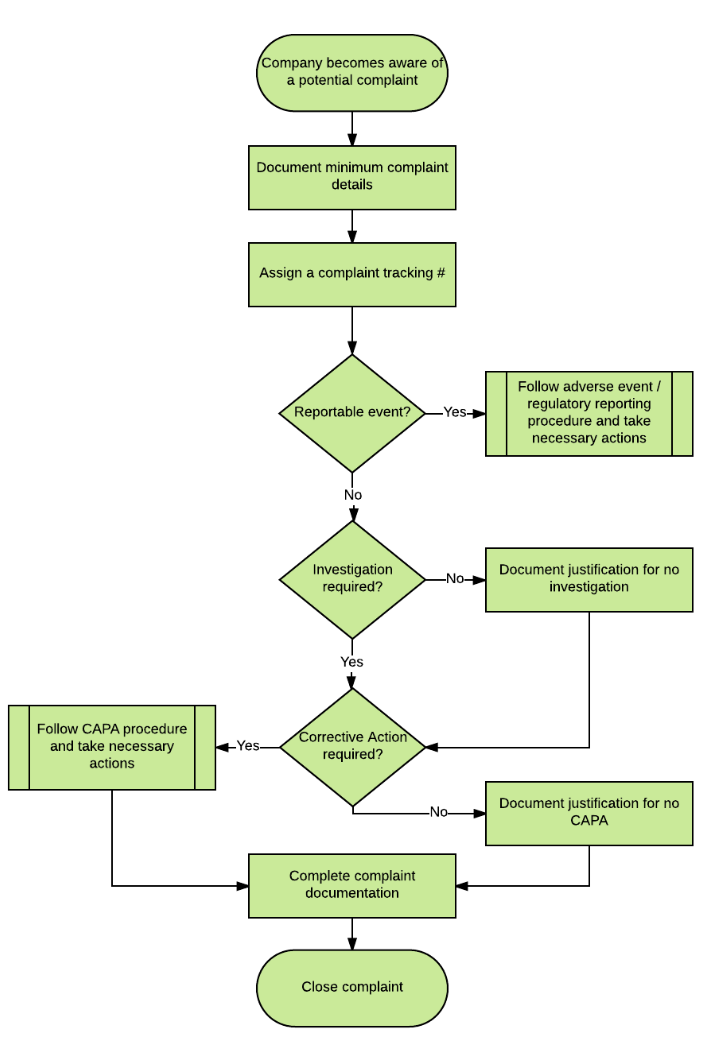
There are three main things you need to determine when receiving a complaint:
- Is this a reportable event that needs to be submitted to regulatory bodies (i.e. FDA)?
- Does this complaint require an investigation?
- Is additional corrective action required?
If your device has caused serious injury or death, or malfunctioned in such a way that it could have done so, you’re obligated to report it regardless of which country the malfunction occurred in.
To answer the second point, reportable complaints always require an investigation under ISO and FDA rules. That investigation must include:
- The name of the device
- Date of the complaint
- Device ID
- Complainant contact information
- Nature of the complaint
- Dates and results of investigation
- Corrective action taken
- Any reply you’ve made to the complainant
CAPAs
For those of us in the industry, we know that Corrective and Preventive Action (CAPA) has been added to the infamous group of four-letter words. CAPAs are heavily scrutinized by regulators and it is important to have a thorough CAPA management process.
Completing a CAPA is often a very document-intensive process, and it’s imperative that you track each investigation with care to ensure CAPA is carried out effectively. Failure to do so could result in a warning letter, or worse, a preventable injury or death. It is also extremely important to close CAPAs in a timely manner and ensure they do not stay open for an extended period of time.
Greenlight Guru’s postmarket workflows is purpose-built to handle all documentation related to postmarket quality events. Workflows are available to handle all aspects of nonconformance, customer feedback/complaint, and CAPA management.
You can monitor quality issue trends in real-time and handle them before they create regulatory problems, assign investigation-related tasks to the relevant teammates, and easily share your automatically-compiled trend data with regulators.
Maintain QMS
In order to effectively maintain your QMS, you will need to conduct a careful analysis of your quality management system (QMS) processes on an ongoing basis. The way you will do this is by holding management reviews.
Management Reviews
Management reviews allow for the examination of your quality management system (QMS) processes and aid the role of executive management in assessing and maintaining your QMS.
Regulators require that you conduct management reviews on an annual basis, but it’s best to do them more frequently.
The reason, as with many other elements of device development, is that simply meeting the minimum requirements tends to result in a drop in quality. People often leave management reviews until the last minute, waiting for the regulatory deadlines to arrive.
A true quality approach to management reviews dictates that you should reduce risk and increase quality by conducting more regular reviews aimed at truly improving your managerial processes, rather than simply meeting the regulator's requirements.
Use key performance indicators (KPIs), as outlined in your Quality Manual, to measure and track your performance. Another reason to conduct regular reviews is the opportunity to set goals between reviews and measure the improvement in your target areas several times within a single year, rather than once a year.
Much like with design reviews, setting an agenda and sticking to it makes your management review process run smoothly.
Internal Audits
Conducting regular internal audits is a great way to make sure that you’re ready for external audits when the time comes.
Here are some best practices when it comes to internally auditing your medical device company.
- Document Your Processes: As with everything, clear documentation makes your life a lot easier. In this case, clear documentation in an internal audit will make it easier for you to figure out where you might be lacking in terms of compliance. Make sure to follow the documentation regulations that apply to your upcoming external audit or audits (ISO, FDA, etc).
- Plan Ahead: This is another best practice that applies to all aspects of medical device development. Planning your internal audits ahead of time reduces the stress of an impending external audit - nothing beats knowing you’re ready for action when the FDA or your NB comes calling. Planning ahead also allows you to take your time and conduct your own audit instead of hiring an outside consultant, which is often not necessary.
- Adopt a Quality Mindset: You shouldn’t just view carrying out internal audits as a check-the-box activity for your procedural requirements. While it’s great to plan an audit and demonstrate to regulators that you’ve followed your procedures, an internal audit is a great opportunity to perform quality gap analyses and improve your company, your product, and as such, the quality of life of your end users.
Manage Regulatory Changes
Just when you feel like you are getting a handle on the regulations that apply to you, it seems like things change. Now more than ever, we are experiencing a whirlwind of regulatory changes in the medical device industry. And we expect that to continue.
We all have been feeling the pain of the changes and headwinds in Europe. Just when we were starting to catch our breath from the ISO 13485 update in 2016, the new EU MDR was enacted. Additionally, updates to ISO 14971 are expected soon.
The changes have led to a major increase in the amount of auditing services being hired, indicating confusion and uncertainty in the industry. It has also led to some concern about the availability of notified bodies.
In the past, it was always assumed that the path to market in Europe was easier than the path to market in the US. That may be changing now, however.
An interesting development in US regulation is the FDA’s growing interest in its ongoing efforts to collaborate with manufacturers.
The FDA’s Center for Devices and Radiological Health (CDRH) launched its Case for Quality program, an initiative designed to allow the regulator to work more closely with manufacturers and guide them through the regulatory process.
Greenlight Guru partnered with FDA in 2018 to promote the initiative - we released a 4-part webinar on the program that I highly recommend.
I believe that manufacturers that embrace the notion of developing a closer relationship with regulators will benefit both parties and, more importantly, the end users.
External Inspections/Audits
While it is hard to find many people that look forward to them, audits and inspections help ensure that you’re delivering a safe, quality product that helps people.
Instead of spending weeks preparing for an audit at the expense of productivity, it’s best to be audit-ready at all times. You might be thinking, “If it’s that simple, why doesn’t everyone do it?”
The truth is that not everyone is prepared and organized. An audit or inspection, when it comes down to it, is simply a measure to ensure that you have all the documentation to prove that you’ve followed your procedures. You need quick and easy access to your procedures, records, and so on.
Audits and inspections go wrong when people simply can't find a particular document, whether it be a procedure, a test report, a drawing, training records - there's a lot to keep track of, and if anything goes missing, the regulators will find out about it.
An easy way to keep track of everything is through the Greenlight Guru’s QMS software. Everything is instantly searchable, and important records don’t go missing from a binder or a hard drive. With everything updating in real-time, you can ensure that everyone’s training is up to date, any open change orders on SOPs are being handled or canceled, and so on.
Greenlight Guru comes with purpose-built Audit Management Software that prepares you for audits well in advance, avoiding the commonly-seen stress of cramming for an audit at the last minute.
FDA Inspections
FDA inspections are often a little more intimidating than other external audits by virtue of being carried out by a law enforcement agency. There is not a lot of personal interaction with your inspector. They don’t even eat lunch with you. It is strictly business.
On average, FDA inspections last around 3-5 days but can vary based on company size and scope. The guidelines for determining whether your company is subject to FDA inspection revolve around the kind of business that you run and also whether the work you do crosses state or international boundaries.
The FDA can inspect your quality system, so it’s important to be prepared to show them that your system is being applied correctly.
If the FDA inspector finds any issues, the findings can be classified in one of two ways. The first is called a 483 observation. You’re quite likely to receive one or more of these following an FDA inspection at some point, and it should be taken seriously.
You’ll receive an observation if the FDA finds something considered potentially problematic during the course of an inspection, and you’ll have 15 days to respond upon receipt of a 483.
Your response should clearly communicate what you intend to do to fix the issues outlined in the observation. We have a comprehensive guide on how to draft a 483 observation response, including cover letter templates and checklists.
A warning letter is an escalation of a 483 observation - it’s less common and much more serious. This could result in hundreds of thousands of dollars in costs, or even being shut down altogether.
You can draft a FDA warning letter response in the same way as you would a 483 observation, and you also must do this in 15 days. If you receive a warning letter, odds are you’ll have received 483 observations on the same issue at the same time - do not treat these as the same.
It’s possible that the issues in the warning letter are not the exact same as in the 483, and they should be handled separately.
ISO Audits
ISO audits are conducted by third-party representatives of the European body called notified bodies. ISO standards are largely derived from European regulations.
If you want to sell devices in international markets like Europe and Canada, you’ll likely need to pass an ISO 13485 audit. The ISO 13485 audit will focus on all areas of your Quality Management System.
Once you have your ISO 13485 certification, you can schedule a technical file to gain CE marking on your device. Successful completion of the technical file audit will result in a CE Certificate.
Both ISO 13485 and CE certificates are valid for three years. The years between are filled with surveillance audits, microbiology audits, unannounced audits, and so on. As you can see, it is important to be prepared for the many different audits you will undergo.
If you fail to meet regulatory standards within the EU single market, you could have your CE Marking suspended or revoked.
A suspension is a temporary measure carried out by your notified body, and the purpose here is to give you the time to correct any issues found during an audit. The Notified Body can then terminate the suspension when you’re found to be in compliance. You can’t process any sales during a suspension, making this a potentially costly experience to go through.
A withdrawal, on the other hand, fully revokes your market approval, meaning you’ll have to go through the entire regulatory process again if this happens.
MDSAP
The Medical Device Single Audit Program (MDSAP) allows companies to be audited once in such a way that they meet compliance with the standard and regulatory requirements of up to five different medical device markets: Australia, Brazil, Canada, Japan and the United States.
The stated goal of the program is to “jointly leverage regulatory resources to manage an efficient, effective, and sustainable single audit program focused on the oversight of medical device manufacturers.”
This has the obvious benefit of allowing you to enter the approval process for multiple markets, and the FDA will accept MDSAP audits as a substitute for FDA inspections. MDSAP audits are done annually and are announced ahead of time. Here is a list of the various MDSAP audit procedures and forms.

![UG-BMDGM_final thoughts_6]() Final Thoughts
Final Thoughts
 Final Thoughts
Final ThoughtsIt’s not easy bringing a medical device to market, and the process can be surprisingly multifaceted and complex for new device developers. However, by implementing a strategic plan from the start, you can eliminate a lot of stress and uncertainty from your process, while increasing the chances for success in bringing your medical device to market.
I hope this guide has given you a strong understanding of the importance of using a good quality management system on your journey to market.
Greenlight Guru Quality is the only QMS software on the market specifically designed to help medical device manufacturers maintain compliance and launch their products. In addition to our solutions, we have a large content library of free online resources, as well as premium resources and features within the software itself to help guide your project to success.
Looking for an end-to-end solution designed to help you through each stage of the go-to-market process? Click here to take a quick tour of Greenlight Guru's Medical Device QMS software →
Complete Guide to Bringing a Medical Device to Market

%20A%20Complete%20Guide%20to%20Bringing%20a%20Medical%20Device%20to%20Market-1.png?width=575&name=(cover)%20A%20Complete%20Guide%20to%20Bringing%20a%20Medical%20Device%20to%20Market-1.png)
%20A%20Complete%20Guide%20to%20Bringing%20a%20Medical%20Device%20to%20Market.png?width=250&name=(cover)%20A%20Complete%20Guide%20to%20Bringing%20a%20Medical%20Device%20to%20Market.png)



