
What is EU MDR?
The European Medical Device Regulation (EU MDR) is a new set of regulations that governs the production and distribution of medical devices in Europe, and compliance with the regulation is mandatory for medical device companies that want to sell their products in the European marketplace.
If your company was already compliant with the Medical Devices Directive (MDD), don't be fooled into complacency - the MDR represents brand new regulations with a lot of changes.
As a starting point for medical device companies, we've created this article where we answer the nine of the most important questions about the new MDR.
We'll cover why the old directive needed an update, the structure of the new MDR document, and some of the latest requirements that all medical device manufacturers and companies need to be aware of.
EU MDR simplified: your key questions answered
FREE RESOURCE: Want to ensure your device is EU MDR compliant? Download your free copy of our EU MDR Gap Analysis Tool to cover your bases.
1. Why did the MDD need an update?There were many reasons the MDD needed to be updated. For instance, when the MDD came into law in 1992, software as a medical device (SaMD) did not yet exist. Software was something that controlled electric machines, and there were no apps that patients could use to monitor their own health.
Demographics have also changed in Europe since 1992, with the population growing older, and there is a growing push for transparency of medical device technical information to the general public.
These combined factors are some of the reasons that the MDD has been replaced with a new regulation that encourages wider compliance with standardized medical device regulations throughout Europe.
2. How is the new EU MDR structured?
The new MDR document is 174 pages in length. It contains a 13-page introduction, followed by 123 articles in 10 chapters (79 pages), and 17 annexes (80 pages). Compared with the MDD, which was 60 pages in length, the new regulations are much longer and more detailed.
In addition to that, there have been published 42 implementing acts, which are used to clarify/implement the MDR and 12 delegating acts, used to modify/amend it.
What's the reason for this much new content? You may not know that the Active Implantable Medical Devices Directive (AIMD) has also been integrated into the new MDR, but besides that, there is still a lot of content that did not exist previously.
3. What are the major thematic changes in the EU Medical Device Regulation?
Compared with its predecessor, the MDD, the new European MDR is less focused on the pre-approval stage of medical device manufacturing, and instead, promotes a lifecycle approach to medical device regulation.
While the old MDD essentially served as a manual for how medical device companies could get their CE marking and get to market, the new regulations encourage policies and procedures that elevate the responsibilities of medical device companies for their products throughout the entire product lifecycle.
The European market comprises 27 member states (not including the United Kingdom) along with others that fall into the European Economic Area (Liechtenstein, Norway, Iceland). The region is inhabited by an aging population of 500 million people, and as populations age, they face greater risks associated with medical device malfunctions and adverse events. This is one of the main reasons that the regulations focus on total product lifecycle, not just pre-market.
4. What devices are covered under the EU MDR?
The EU Medical Device Regulation (EU MDR) defines the term "medical device" as an "instrument, apparatus, appliance, software, implant, reagent, material, or other article" that is used for any of the following:
-
Diagnosis, prevention, monitoring, treatment or alleviation of disease, disability, or injury, but not for disability or injury prevention
-
Investigation, replacement, or modification of an anatomical, physiological, or pathological process
-
Providing data via in-vitro examination of samples derived from a human body
This definition covers a broad range of existing devices, but that's not all. The MDR newly specifies certain types of products that need to obtain a CE marking, including products used to clean, disinfect, or sterilize medical devices, and devices used to control and support conception, whether through pharmacological, immunological, or metabolic means.
It is important to consult the regulations directly to determine whether your medical device is covered under the MDR. Even so, you may be spared some of the hardships of compliance depending on your device classification. Under the MDR, Class I devices are exempt from being required to have their QMS audited by a third-party auditor (notified body).
5. What devices are covered under Annex XVI of the MDR?
The new MDR contains a total of 16 Annex sections, and the one creating the most buzz is Annex XVI. This section mandates that certain devices are covered under the MDR - ones that may not have previously been considered medical devices. As a result, some companies are being subjected to medical device compliance regulations for the first time.
Annex XVI mandates that the following groups of products comply with the requirements of the MDR:
-
Contact lenses and other products used in or on the eye (eye drops and cosmetic contact lenses would be included here)
-
Products introduced into the body via surgically invasive means to modify the anatomy (silicone breast implants would now qualify here)
-
Products and substances used for facial or other subcutaneous fillings
-
Equipment used for liposuction, lipolysis, or lipoplasty
-
High-intensity radiation equipment used for tattoo and hair removal
-
Equipment using electrical or magnetic currents to stimulate the brain
Annex XVI plugs a lot of gaps that existed in the previous MDD, specifically imposing itself on devices used in procedures that escaped regulation because they were cosmetic and not medical in nature.
6. Does EU MDR require enhanced device traceability and technical documentation?
Yes, it does. The new MDR includes a mandate for Unique Device Identification (UDI) that is intended to facilitate the traceability of all medical devices sold in the region. Devices must be marked with a device identifier (DI) and each batch or production series of the product will be marked with a production identifier (PI).
The MDR also introduces new databases for clinical investigations, product registration, and post-market surveillance. The EUDAMED database will function as part of a system of several databases that allows Notified Bodies, medical device companies, consumers, regulators, and other stakeholders to access the latest data on medical devices for sale in Europe.
7. How Will the MDR Affect CE Markings?
The new MDR significantly impacts the steps to not only obtaining a CE marking, but maintaining it as well.
The CE marking on a medical device allows it to be freely traded in the EEA, and legislators are anxious to ensure the device meets all regions’ standards for patient safety and environmental protection. As a result, the process of receiving the CE marking is far more rigorous than under the Medical Device Directive of years past.
Here are some of the major changes in CE marking for medical devices:
-
Classification. Rather than depending on predicate devices or a decision tree, device classification under MDR is determined by intended use.
-
Person Responsible for Compliance. Article 15 of MDR requires the appointment of a Person Responsible for Compliance (PRRC), a designated employee with regulatory experience as defined in the regulation, as well as experience with quality management systems or regulatory affairs.
-
Authorized Representative. Any manufacturers not established in the EU must appoint an Authorized Representative before obtaining a CE marking. Authorized Representatives must be operating in the EU, and will remain the primary liaison between the manufacturers, notified bodies, and national competent authorities. The contact information of the Authorized Representative must also be displayed on all packaging and labeling.
-
Notified Body Review. This process will have far more scrutiny, with three rounds of questions and require a significant investment in resources and time. If your device is approved, your CE marking certificate will be good for up to five years.
-
Maintaining CE marking. Though the CE marking is valid for five years, manufacturers are still required to undergo random audits from notified bodies, keep ongoing technical documentation, and perform extensive post-market surveillance.
8. How does EU MDR affect clinical evaluation requirements?
In order to receive a CE marking, MDR now requires manufacturers to first submit clinical evaluations in the form of a Clinical Evaluation Report (CER). CERs provide essential data on a device's safety and performance and are used in the conformity assessment process. As such, you'll need to include the report in the CE technical file.
In addition, device manufacturers can expect the following systemic changes in MDR's approach to clinical evaluations:
-
Reclassification. In vitro diagnostic devices that are classified as "General" (meaning they do not require approval from a notified body) may be reclassified to "Annex II List A" (meaning they would require both approval from a notified body, as well as additional audits and reviews).
-
Scrutiny Process. Under the new scrutiny process, authorities may choose to review technical documentation again before approving high-risk devices. This could impact how long it takes to get approval.
-
Comparative Evaluations. MDR includes more stringent requirements in the comparative evaluation process. In order to better demonstrate product safety and performance, you’ll need to produce, collect, and analyze a much larger amount of data.
9. How does the new MDR affect my QMS?
Under MDR, any and all previous “directives” will no longer exist or be applicable to quality systems. Instead, they will have a new name and carry a much more substantial meaning -- Regulations. Simple translation is that these changes are now going to be considered laws—similar to FDA medical device regulations.
Manufacturers shall follow what is now known as “General Obligations” to ensure compliance for their quality management systems (QMS). This comprehensive list can be found in the new MDR starting from Article X.
As stated in the MDR, manufacturers shall establish, document and implement a quality system, and maintain its effectiveness throughout the entire device lifecycle. There’s also a strong emphasis placed on the role management plays in QMS activities such as, document storage, post-market surveillance, and risk assessments of new and existing devices.
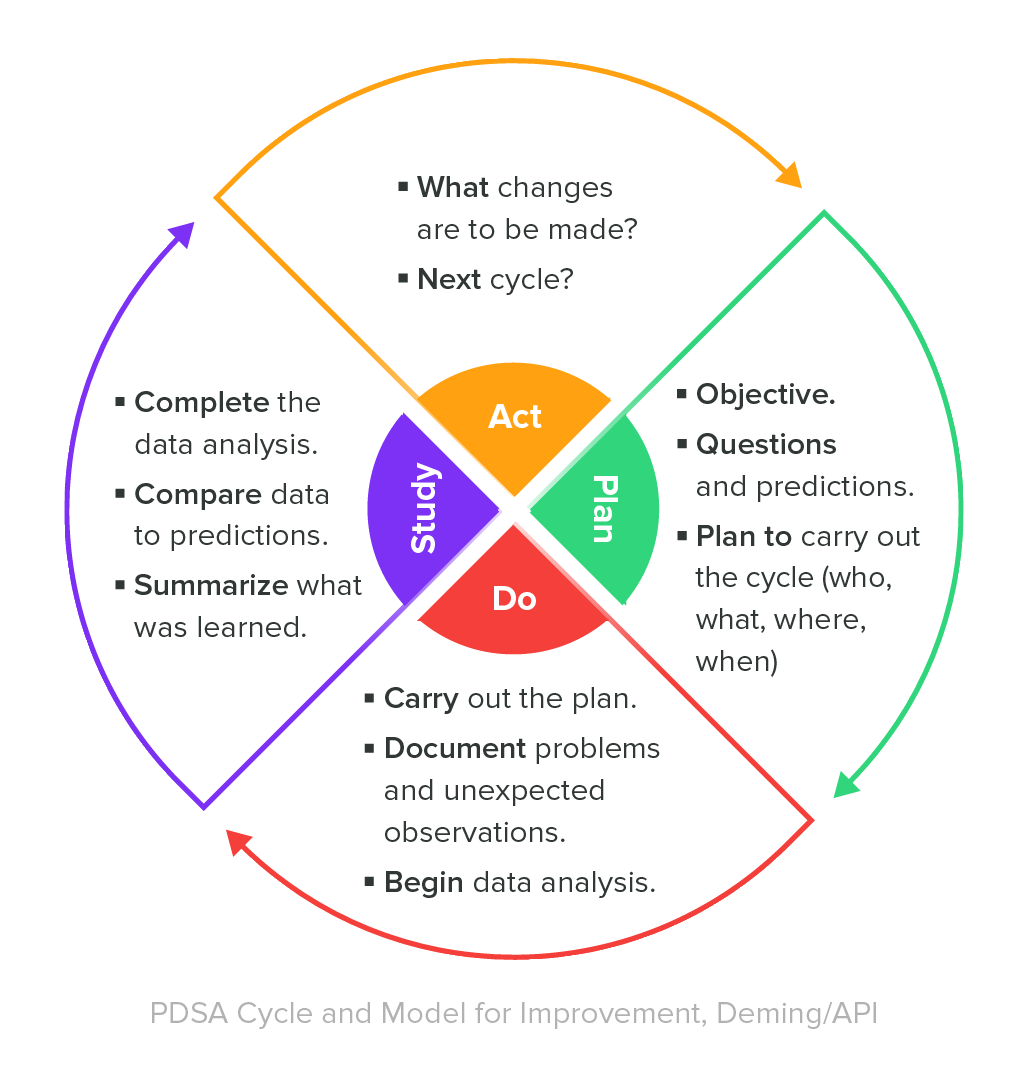
During QMS implementation, manufacturers must include documentation in the form of a quality manual and written policies/procedures, including:
-
Quality objectives.
-
Organization of the business.
-
Procedures/techniques used for design control, verification, validation, and review of the device(s).
-
Quality assurance and verification procedures during the manufacturing stage.
-
Relevant tests and trials.
In the event a manufacturer plans to implement a new quality system, they must submit “an application for assessment of its quality system with a notified body” (excluding Class I devices) and the documentation of the application must include:
-
Manufacturer’s current QMS and the procedures used to ensure adequacy and efficacy.
-
Manufacturer’s PMS system and clinical evaluation plan, along with descriptions of both of those procedures to ensure these will be kept up-to-date.
-
Current procedures being used to meet both QMS and MDR / IVDR regulations must be documented along with descriptions of each.
FREE RESOURCE: Click here to download your copy of our EU MDR Gap Analysis Tool.
Focus on the future of European MDR with Greenlight Guru
Much like the MedTech industry, the EU Medical Device Regulation is a living, still-evolving thing.
Even today, new guidelines are frequently published clarifying the legislation’s push for greater standardization, stronger post-market surveillance requirements, process-oriented risk management, and a lifecycle approach to device management.
To keep up with the pace, you need a future-proof solution that is built to help you ensure compliance throughout the entire product lifecycle. That’s where Greenlight Guru comes in.
Greenlight Guru's Quality Management Software is built to accelerate your product lifecycle from ideation to commercialization to postmarket surveillance. Get a free demo of our QMS software, today!
For clinical teams, it's important to have a validated clinical data management system that streamlines data collection from clinical investigations. Greenlight Guru Clinical (formerly known as SMART-TRIAL) is the only MedTech-specific electronic data capture system that empowers clinical teams to capture, collect and manage clinical data from clinical investigations in a compliant and secure environment. See it in action, book a demo.
FREE RESOURCE:
EU MDR Gap Analysis Tool
EU MDR Gap Analysis Tool






