
What I am about to share with you is a guide to medical device regulatory classification. In this guide, I will provide you with a step-by-step approach to determining how your medical device will be classified by U.S. FDA, the European Commission, and Health Canada. Understanding the different medical device classes will be invaluable to bringing new products to market.
Medical device regulatory classification
The rules that apply to your medical device depend on how your product is classified by the regulatory agencies. Each regulatory agency has defined several different medical device classes.
The classifications are, for the most part or as a general rule, related to the perceived risk of the product type.
Medical device manufacturers selling internationally need to familiarize themselves with the applicable regulations of those markets. This is easier said than done and can be a challenge for most manufacturers. The US has its set of rules, while Canada adheres to another, and Europe another one still.
Fortunately, there are many parallels between international medical device regulations and standards. This guide is designed to show you how to classify your device in different markets around the world.

Why does medical device classification matter?
Knowing how your medical device is classified matters for the following reasons:
- Product classification will determine what you have to do before you can sell your product.
- Product classification will help you establish requirements during the product development phase, specifically design controls.
- Product classification is an important component in determining how much it will cost to bring your device to market and give you some idea of how long it will take.
Because of this, I’m going to provide you with a little bit of guidance to better understand what to do and how to do it.
Note, the information I’m about to provide is intended to help educate you on medical device regulatory classification and what is required for your medical device.
The following content is not a comprehensive guide to regulatory submissions, yet should give you some basic guidance and direction on identifying how to establish path to market.
I’ll stick to the “big 3” you should know when it comes to medical device classification:
- U.S. Food & Drug Administration, Center for Devices & Radiological Health (FDA CDRH)
- European Commission
- Health Canada

Medical Device Classification in the United States - FDA CDRH
In the United States, medical devices are regulated by the Food & Drug Administration, or FDA. The specific branch within the FDA is the Center for Devices & Radiological Health (CDRH).
The mission of CDRH is to protect and promote public health. In other words, ensure medical devices are safe. In the U.S., medical devices are either Class 1, Class 2, or Class 3 (notated as Class I, Class II and Class III by the FDA).
The FDA medical device classes are based primarily on the risk the device poses.
Class I medical devices are generally deemed low risk. Class II medical devices are associated with a higher risk level than class I devices, but less than that of class III devices. Class III medical devices are seen as the highest risk. The types of controls required is dependent on your product’s classification.
Classification is directly related to intended use and indications for use. The distinction between these terms is a bit confusing.
-
Intended Use is the general purpose of the medical device or its function (what you “claim” the medical device does).
-
Indications for Use describe the disease or condition the medical device will diagnose, treat, prevent, cure, or mitigate, including a description of the target patient population.
Remember, the intended use and indications for use of your medical device convey the reasons you developed this new medical device in the first place.
How to find the applicable regulatory classification for your medical device
Once you define intended use and indications for use, now you need to find the possible regulations and product codes. Tracking down regulatory classification for your product via FDA takes a little bit of time and perseverance.
Without boring you with too many details, FDA has established several general categories based on the medical specialty in CFR Title 21 - Food and Drugs: Parts 862 to 892.
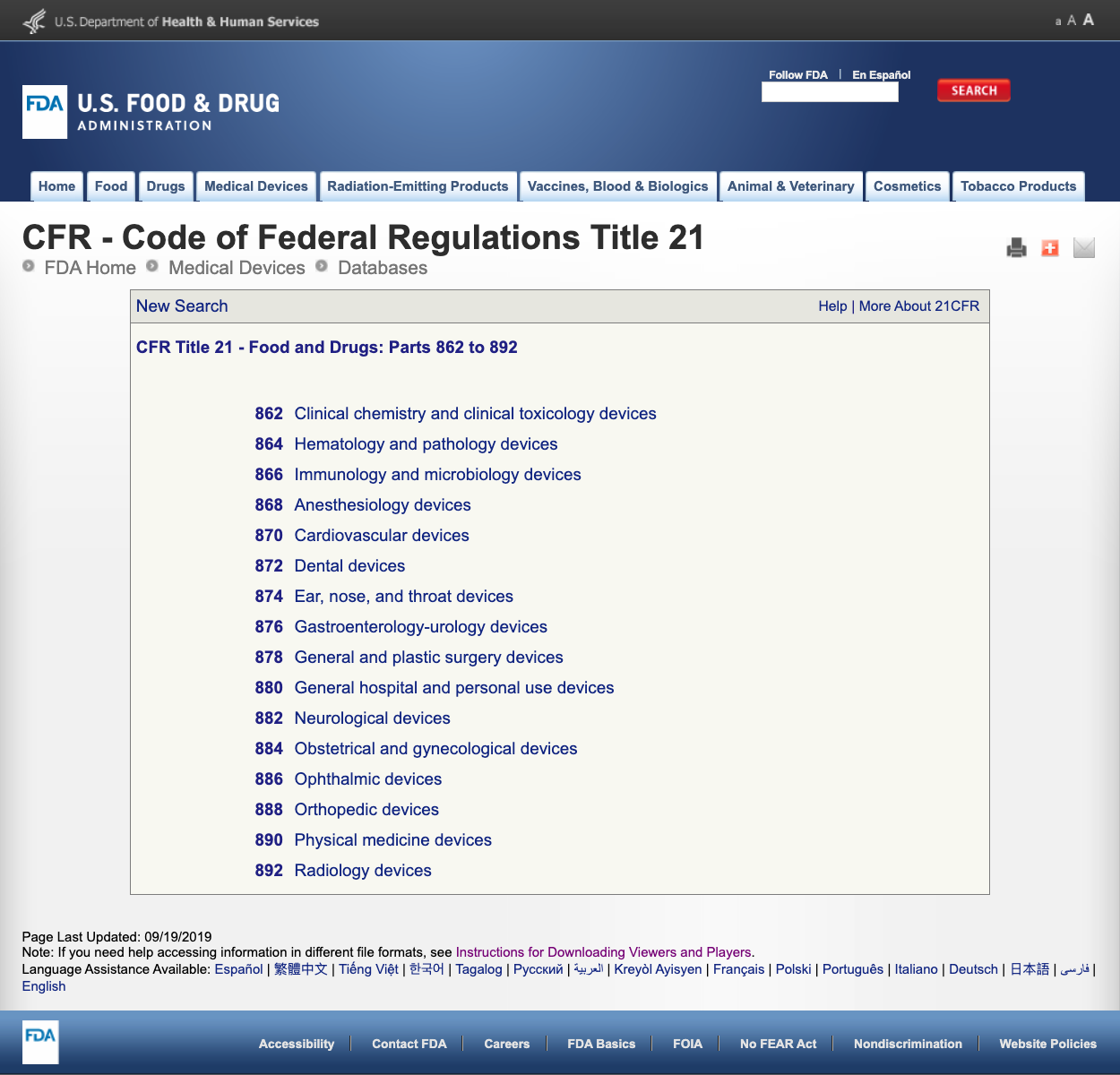
When you find the possible categories and click on the FDA regulation number, the list of possibilities suddenly seems endless. Here is a partial view of the options for Part 870 Cardiovascular devices:

When you find a regulation that appears to be a possible fit, you can click on the link and get more details to make a determination.
For example, if I think my device fits in Part 870.1250 - Percutaneous catheter, I click the link and get this information:
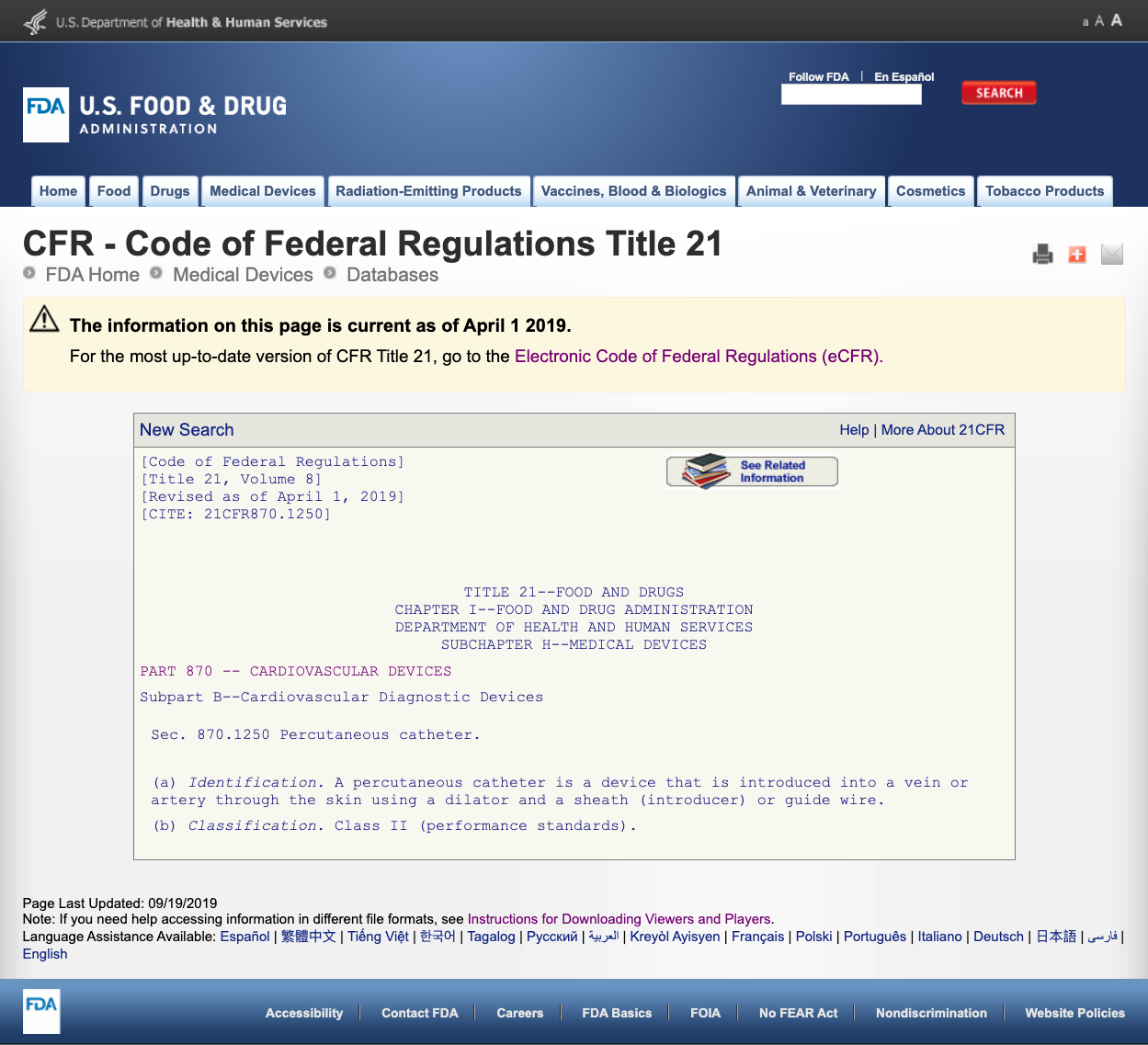
The details provided give me some idea if my intended use and indications for use align with this specific regulation. I also discover the FDA device classification.
In this example, I learn that my product is a Class II medical device (performance standards), which means I will need to submit a 510(k) to FDA prior to getting market clearance. I share more about types of FDA submissions further on in this guide.
How to find the product codes applicable to your device classification
Finding the applicable regulation for your medical device and classification is the first part. Now you need to find the applicable product codes. Here’s how:
Go to the FDA Product Classification Database and type in the regulation number you found. If you find more than one possibility, then you will need to repeat this process for each.
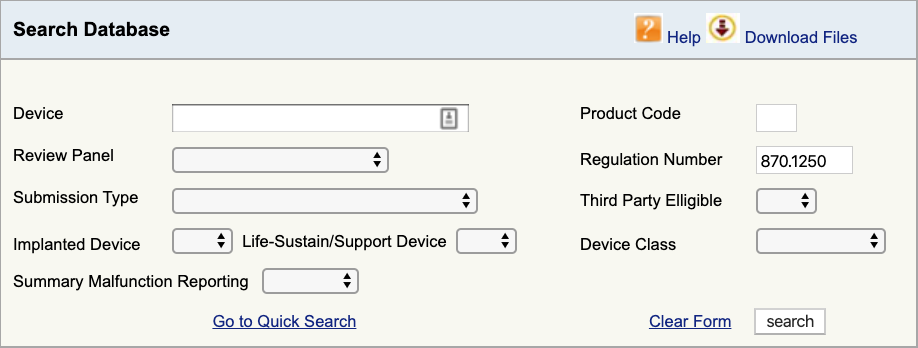
When you click “search”, you will get a list of possible product codes.
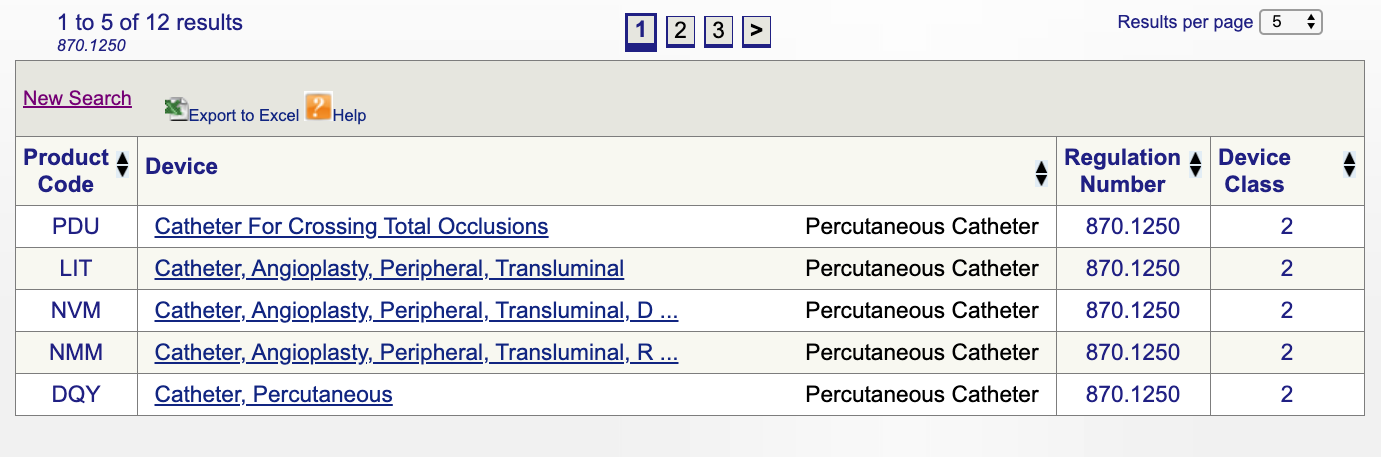
You can then review each individual code to determine the best option for your product by clicking on each code.
How medical device classes determine pathways to market in the U.S.
Knowing the applicable regulation and product code (as described above) is necessary for you to determine the classification of your medical device. Once you have this information, you will now be able to determine the pathway to get your product registered with FDA.
FDA defines three regulatory controls:
-
Class I medical device (low to moderate risk): General Controls
-
Class 1 medical devices are notated as Class I devices by the FDA and are subject to only general controls. Most class 1 devices are not required to submit a premarket notification.
-
-
Class II medical device (moderate to high risk): General Controls and Special Controls
-
Class 2 medical devices, notated as class II devices by FDA, are subject to the same general controls as class 1 devices, in addition to special controls such as performance standards, postmarket surveillance, and premarket notification requirements, most commonly known as a 510(k) submission, for FDA clearance to legally market the device.
-
-
Class III medical device (high risk): General Controls and Premarket Approval (PMA)
-
Class 3 medical devices, notated as class III devices by FDA, are subject to both general and special controls, as well as Premarket Approval (PMA) from FDA in order to be legally marketed in the US.
-
Let me boil it down to this:
If you find your product is “exempt,” then only general controls apply and no formal FDA submission is required. You do, however, need to register your establishment with FDA and then list the product.
If you find your product requires special controls, this means you will have to prepare a 510(k) submission to FDA and receive clearance before going to market. After that, you need to register your establishment and list the product.
If you find your product requires premarket approval, this means you will have to follow the FDA PMA process to receive approval before going to market.
.png)

Medical device classification in Europe - European Commission (EC)
The regulations for a medical device in the European Union (EU) are established through EU MDR 2017/745 by the European Commission (EC).
On 26 May 2021, EU MDR became applicable in the European Union. The regulation amends Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repeals Council Directives 90/385/EEC and 93/42/EEC.
The path to market in Europe is to obtain a CE marking. The requirements to obtain CE-marking are based on the EU system of medical device classes. The European Union’s medical device regulation (EU MDR) includes the necessary information to determine your device class.
You will need to determine if your medical device is:
- Non-Invasive. Any device which does not penetrate the body through an orifice or the surface of the body. These devices are typically Class I; however, certain rules and exceptions apply that could make them Class II devices or higher.
- Invasive. Any device which, in whole or in part, penetrates inside the body, either through a body orifice or through the surface of the body.
- Active. Any device whose operation depends on a source of energy other than that generated by the human body for that purpose, or by gravity, and which acts by changing the density of or converting that energy.
For each of the broad categories, there are certain rules which apply, outlined in Annex VIII of the new medical device regulation. These categories coupled with the duration for use make determining classification fairly straightforward.
For example, a device in continuous use for under 60 minutes is considered transient duration, 60 minutes to 30 days is considered short-term, and over 30 days is considered long-term.
With that in mind, to determine the EU classification of your device, we can use the percutaneous catheter example used earlier in this guide for FDA classification.
Let’s say I determine my medical device fits into the “invasive” category; this narrows my search down to Rules 5, 6, 7, and 8.
I can then narrow the rules down further since I know my medical device is short-term because it is used for a period greater than 24 hours and less than 30 days.
From there, I am able to determine that Rule 7 is most applicable to the classification of my device.
5.3. Rule 7
All surgically invasive devices intended for short-term use are classified as class la unless they:
-
-
are intended specifically to control, diagnose, monitor or correct a defect of the heart or of the central circulatory system through direct contact with those parts of the body, in which case they are classified as class III;
-
-
-
are intended specifically for use in direct contact with the heart or central circulatory system or the central nervous system, in which case they are classified as class III;
-
-
-
are intended to supply energy in the form of ionizing radiation in which case they are classified as class lIb; have a biological effect or are wholly or mainly absorbed in which case they are classified as class Ill;
-
-
-
are intended to undergo chemical change in the body in which case they are classified as class lib, except if the devices are placed in the teeth; or
-
-
-
are intended to administer medicines, in which case they are classified as class IIb.
-
Since my percutaneous catheter will administer medicines, I can confirm that my medical device is considered a Class IIb medical device in the European marketplace.
How medical device classes determine pathways to market in Europe
The European Union has a similar product classification system as the FDA medical device classes:
- Class I medical device
- Class IIa medical device
- Class IIb medical device
- Class III medical device
All medical devices to be sold in the European Union must first obtain a CE marking, a process which requires extensive technical documentation. All medical device classes in the EU require working with a Notified Body, except for those which are Class I and can be self-certified.
Companies will also need to work with an Authorized Representative to take care of product registration in Europe.
Medical Device Classification in Canada - Health Canada
The medical devices regulations in Canada are established by the Government of Canada and regulated by Health Canada.
Like the U.S. and EU, to sell into the Canadian marketplace, you must first determine the medical device classification under Canada’s regulation.
Similar to the requirements outlined in EU MDR, Health Canada provides a fairly straightforward and easy to follow Guidance on the Risk based Classification System for Non-In Vitro Diagnostic Devices for medical device manufacturers to use when selling into this market.
Health Canada defines four groups of non-in vitro diagnostic medical devices:
- Invasive Devices (Rules 1 - 3)
- Non-Invasive Devices (Rules 4 - 7)
- Active Devices (Rules 8 - 12)
-
Special Rules (Rules 13 - 16)
For each of the broad categories, there are a set of rules which apply. These rules are what manufacturers should follow in order to determine the risk classification of their device.
I’ll use the percutaneous catheter example once more in the case of marketing such a device in Canada.
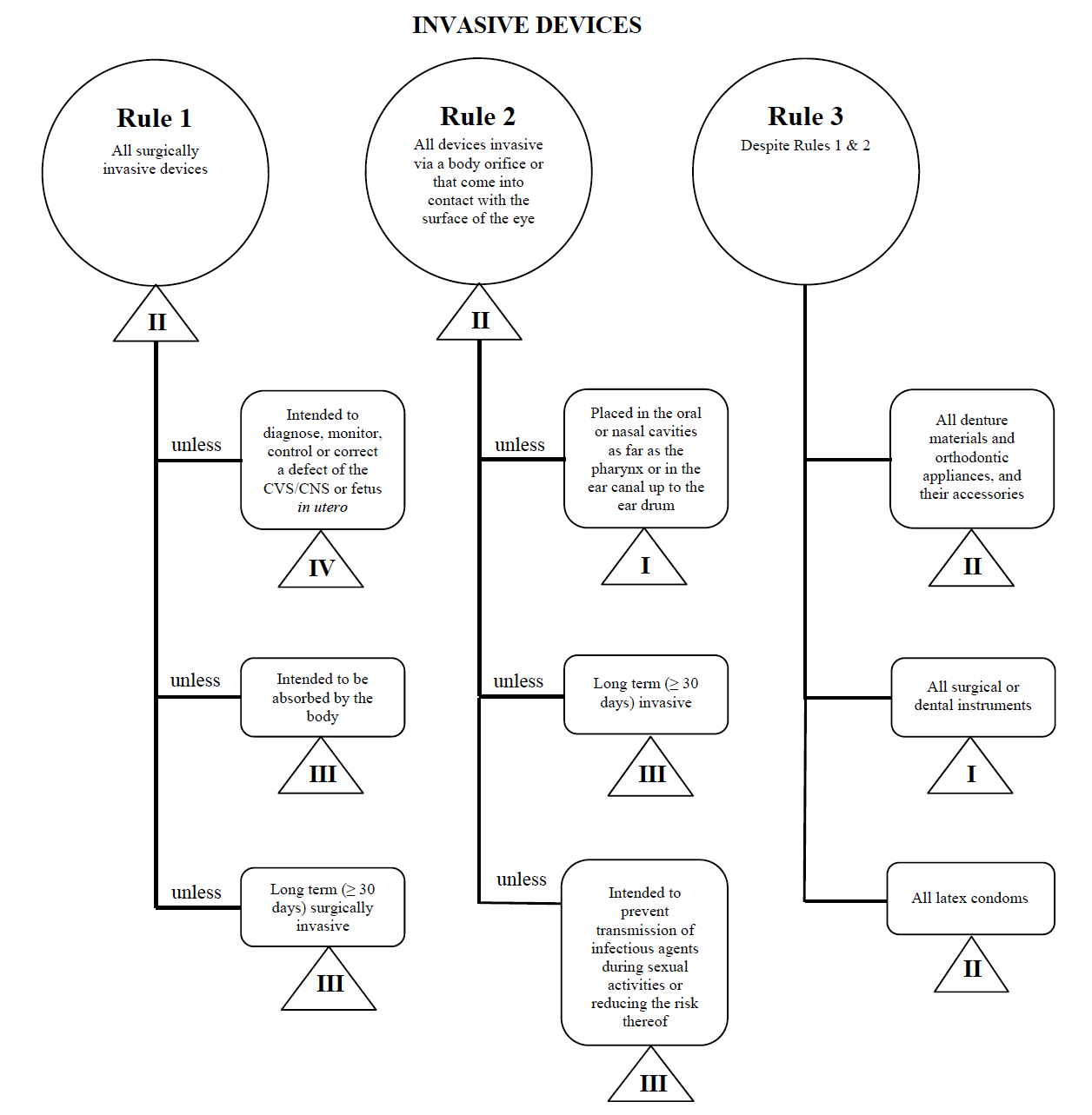
Source: Guidance on the Risk based Classification System for Non-In Vitro Diagnostic Devices
I determine my medical device fits into the “invasive” category, narrowing my search down to Rules 1, 2, and 3.
After reviewing the options, I determine that Rule 1 applies.
Based on my intended use, my medical device is considered Class II in Canada.
How medical device classes determine pathways to market in Canada
There are four levels of medical device classes in Canada:
- Class I medical device
- Class II medical device
- Class III medical device
- Class IV medical device
Prior to going to market in Canada, you must first apply for a medical device license. Class I medical devices do not require a license. Manufacturers can reference the Health Canada guidance document, which walks you through this process.
Manufacturers of Class III and Class IV medical devices can receive their license by submitting a premarket application, in either the ToC or Health Canada formats, for entering the Canadian market.
You will also need to obtain ISO 13485 certification with MDSAP.
Update to Health Canada regulations: MDSAP
In the Canadian market, all devices categorized as Class II or higher must be part of the Medical Device Single Audit Program (MDSAP).
These manufacturers must undergo and pass a full audit of their quality management system (QMS) through the program in Canada. There are currently 6 regions around the world that participate in MDSAP, including Canada, U.S., Japan, Brazil, and Australia.
Aside from those manufacturers required by Health Canada to participate in the program, participation in MDSAP is optional for manufacturers.
To gain MDSAP certification, medical device manufacturers must complete the following three steps:
- Application and review
- Off-site documentation audit
- On-site audit
Device makers selling into Canada will be subjected to annual reviews, with a recertification audit every third year. Compliance with the Medical Device Single Audit Program is based on meeting the guidelines from the ISO 13485 standard on quality management systems for medical devices.
We recommend training your product and quality and regulatory teams in the applicable MDSAP requirements to streamline your application.
Our free gap assessment tool for MDSAP and ISO 13485 helps device makers assess the QMS guidelines from the ISO standard alongside the requirements of Health Canada auditing organizations (AO) and other regions participating in the programme.

Streamline medical device classification and get to market 3x faster with Greenlight Guru
Whether you are submitting to the EU, FDA, Health Canada, or others, your path to market and steps to success are determined by your classification and the requirements that follow.
Regardless of the classification of your device, it's imperative that you follow the regulatory guidelines that apply in each market. Ensuring compliance is a key aspect of quality management that will ultimately decide the fate of your device and company as a whole.
At Greenlight Guru, we built our QMS software for MedTech companies to comply with all key standards and regulations. That's why our QMS comes pre-validated per FDA and ISO best practices—so you don't have to worry about customization for compliance.
Ready to learn more? Get your free, personalized demo of Greenlight Guru today!
Free eBook: Medical Device Classification Guide

%20Device%20Classification.png?width=180&name=(cover)%20Device%20Classification.png)
%20Device%20Classification.png?width=250&height=324&name=(cover)%20Device%20Classification.png)



