
In the US, the Food and Drug Administration (FDA) is the federal agency tasked with regulating the medical device market and ensuring the safety and effectiveness of all devices for patients.
FDA classifies medical devices by risk into three categories, Class I, Class II, and Class III, with Class III medical devices as the highest risk class.
FDA defines a Class III medical device as “one that supports or sustains human life or is of substantial importance in preventing impairment of human health or presents a potential, unreasonable risk of illness or injury.”
Given the significant risk inherent in Class III devices, and the often critical nature of their role in medicine, Class III devices are subject to the most stringent regulatory controls. In this article, I’ll cover device classification, regulatory pathways, and more for Class III medical devices.
BONUS RESOURCE: Click here to download a free, customizable PMA Inspection Checklist.
How does FDA categorize Class III medical devices?
FDA assigns a device to a risk class based on the level of regulatory controls required to ensure that the device is safe and effective.
-
Class I medical devices (low to moderate risk) are subject to general controls
-
Class II medical devices (moderate to high risk) are subject to both general controls and Special Controls
-
Class III medical devices (high risk) are subject to general controls and premarket approval
As you can see, all devices are subject to general controls, though Class III devices are the only class subject to the premarket approval (PMA) process, as the FDA deems that general and special controls alone are not enough to assure their safety and effectiveness.
While devices are classified based on risk they pose and the regulatory controls necessary to ensure safety and effectiveness, classification is also dependent on both the intended use of the device and its indications for use.
The classic example is a scalpel, which has an intended use of cutting tissue and is generally a Class I device. However, if a manufacturer were to choose a more specialized indication for use—such as making an incision in the cornea—the risk associated with the device would increase. In that case, it would become a Class III medical device.
What are some examples of Class III medical devices?
Class III medical devices exist in a wide range of medical specialities, and it’s not always obvious that a device is deemed to be high-risk.
For example, here are several examples of Class III medical devices:
-
Pacemakers
-
Defibrillators
-
Implanted prosthetics
-
Cochlear implants
-
Breast implants
-
Intraocular lenses
-
Renal stents
-
Software as a Medical Device (SaMD) that meets the FDA’s criteria for a Class III device
How do you determine if you have a Class III medical device?
You will be able to determine the classification of most medical devices by finding the matching description of the device in 21 CFR Parts 862-892. Within these Parts, FDA has described and organized more than 1,700 distinct types of devices and organized them by medical specialty, e.g., Part 872 covers Dental Devices.
You can also search the FDA’s classification database using part of the device’s name. Once you find the product that corresponds with your device, you will also be able to see its risk class. For example, a catheter balloon repair kit is a Class III cardiovascular device. Here is how it appears in the classification database:
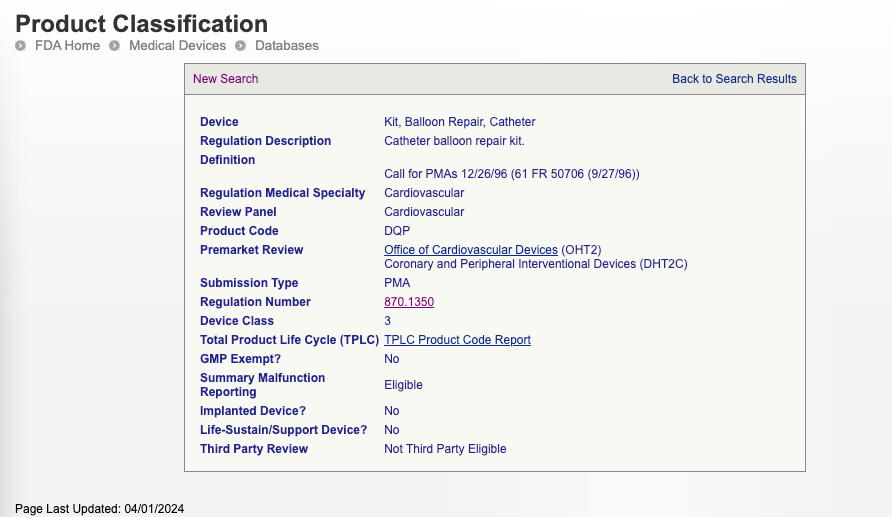
For the most part, you should be able to determine your device’s risk class using these methods, but if you would like a formal determination of device class from FDA, you can submit a 513(g) request.
What is the premarket approval process for Class III medical devices?
All Class III medical devices are subject to premarket approval as part of their regulatory controls. If your device is Class III, you’ll be required to submit a PMA application and must receive FDA approval of said application before marketing your device in the US.
You can find the full requirements for the premarket approval process in 21 CFR Part 814, and for a more streamlined version, check out our guide to the PMA submission process.
The PMA process is lengthy and requires extensive scientific review of your device and the clinical evidence regarding its safety and effectiveness. It’s important here to remember that Class III medical devices require some level of clinical investigation to obtain the appropriate clinical data on their safety and effectiveness.
One of the largest sections in your PMA application will be devoted to clinical and non-clinical investigations of your device, and irregularities in your clinical trials are grounds for denial of approval by FDA.
Additionally, even after Class III medical device has been approved by FDA, the agency will still require post-approval activities, which may include post-approval studies to ensure the long-term safety and effectiveness of the device.
That’s why it’s a good idea to use a modern, compliant electronic data collection (EDC) system like Greenlight Guru Clinical for your clinical data collection. Our comprehensive EDC solution helps you stay compliant from day one and ensures traceability and security for your clinical data.
BONUS RESOURCE: Click here to download a free, customizable PMA Inspection Checklist.
Greenlight Guru helps you stay compliant throughout the lifecycle of your Class III medical device
It takes an enormous amount of time and effort to get a Class III device to market in the US, but these are some of the most important and impactful medical devices for many patients. At Greenlight Guru, our mission is to improve the quality of life by helping MedTech companies deliver the safest, most effective medical devices to patients faster.
To do that, we’ve built our MedTech Suite—QMS, product development, and clinical data solutions—specifically for companies in our industry. These purpose-built solutions are all aimed at reducing the time MedTech companies spend on manual, administrative work, and increasing their ability to innovate and build products that will change lives.
If you’re ready to see how our MedTech-specific solutions can help you stay compliant throughout the lifecycle of your Class III medical device, then get your free demo of Greenlight Guru’s software today →
Premarket Approval Inspection Checklist






