-1.png?width=750&height=375&name=Ultimate%20Guide%20to%20Software%20as%20a%20Medical%20Device%20(SaMD)-1.png)
Software as a Medical Device (SaMD) is software intended for one or more medical purposes that performs those purposes without being part of a physical hardware medical device. SaMD may diagnose, monitor, analyze, or inform treatment decisions using medical data, patient data, imaging, signals, or other health information.
The key distinction is independence from the hardware device. Software that controls or drives a hardware medical device is typically software in a medical device (SiMD), while SaMD performs a medical function on its own. SaMD teams need to consider intended use, risk classification, software lifecycle processes, cybersecurity, clinical evaluation, AI/ML controls where applicable, and regulatory expectations from frameworks such as FDA, EU MDR, IMDRF, IEC 62304, ISO 13485, and ISO 14971.
IEC 62304, ISO 14971, andISO 13485 all apply to these devices, and for teams coming from a software background, the regulatory reality around SaMD is often a surprise. We built this guide to help anyone looking to get the full picture about SaMD classification, global regulations, software development requirements, cybersecurity, and more. You'll find that everything you need to build foundational knowledge about SaMD, or brush up on some specific points, is in this guide.
SaMD Overview
- SaMD is software that performs a medical purpose independently from hardware.
- SaMD is still regulated as a medical device when it meets the applicable device definition in the market where it will be sold.
- Intended use is the starting point for determining whether a product is SaMD.
- SaMD is different from SiMD. SaMD performs the medical function on its own; SiMD is software that is part of, or controls, a hardware medical device.
- IEC 62304, ISO 14971, and ISO 13485 are all relevant to SaMD manufacturers.
- Cybersecurity, SOUP management, SBOMs, validation, and postmarket monitoring should be addressed throughout the product lifecycle.
SaMD vs SiMD vs Health Software
Not every healthcare software product is SaMD. The distinction usually depends on the software’s intended medical purpose and whether it performs that purpose independently from a hardware device.
| Software Type | Concise definition | Example | Regulatory Implication |
| SaMD | Software that performs a medical purpose on its own, without being part of a hardware medical device. | Standalone software that analyzes medical images to support diagnosis. | May be regulated as a medical device based on intended use and risk. |
| SiMD | Software that is part of, controls, or drives a physical medical device. | Firmware that controls an infusion pump or imaging device. | Regulated as part of the hardware device’s safety and performance. |
| General health software | Software that supports wellness, administration, education, or non-medical health management without a regulated medical purpose. | A general wellness app that tracks habits without diagnosing or treating disease. | May fall outside medical device regulation depending on claims and intended use. |
Let’s take it from the top.
|
|
|
|
|
|
|
|
|
|
|
|






.png)
Table of Contents

![SaMD - Software as a Medical Device - Chapter One]() What is software as a medical device (SaMD)?
What is software as a medical device (SaMD)?
 What is software as a medical device (SaMD)?
What is software as a medical device (SaMD)?Software as a medical device or SaMD is “software intended for one or more medical purposes that perform those purposes without being part of a hardware medical device,” as defined by the International Medical Device Regulators Forum (IMDRF).
FDA defines SaMD as “Software that meets the definition of a device in 181 section 201(h) of the FD&C Act and is intended to be used for one or more medical purposes without being part of a hardware device.”
While these definitions are a great starting point, there’s plenty of nuance around what exactly SaMD is and how you know if your product is SaMD. So, let’s take a closer look at what software as a medical device is, what it isn’t, and how you can figure out if your product fits the definition.
How do I know if my product is SaMD?
The key starting point is intended use. Before you can determine whether your software is SaMD, you need to clearly define what the software does, who it is for, and what clinical or medical purpose it is intended to serve.
FDA is a member of the IMDRF, and it’s clear that both definitions of SaMD share a similarity in structure. According to both FDA and IMDRF definitions, there are two points that need to be fulfilled for software to achieve the status of SaMD.
To start, we need to consider whether the software can be characterized as a medical device at all. The IMDRF simply states that it must be “intended for one or more medical purposes”, while FDA specifically references the definition of a device in 181 section 201(h) of the FD&C Act, which states that a device is:
An instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory which is:
-
-
recognized in the official National Formulary, or the United States Pharmacopoeia, or any supplement to them,
-
intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or
-
intended to affect the structure or any function of the body of man or other animals, and which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and
-
which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of its primary intended purposes. The term "device" does not include software functions excluded pursuant to section 520(o).
To appropriately use this definition, you need to define your product’s intended use and its indications for use. As a refresher:
-
Intended use is the purpose of your device. It’s what your device will be used for.
-
Indications for use are the diseases or conditions that your device will diagnose, treat, prevent, cure, or mitigate. Indications for use describe who your device will be used on and why.
Once you’ve defined these uses, points two and three in the FDA’s definition of a medical device should make it fairly clear whether your product will be regulated as a medical device.
If you plan on marketing your software product in the US, I highly encourage you to read carefully through this FDA guidance on Policy for Device Software Functions and Mobile Medical Applications. This guidance offers clear stances on what software functions FDA considers to be a medical device, as well as those it does not consider to be medical devices, or those which it does not regulate as such.
However, if you’re still not sure if your product is a medical device, your best bet is to contact FDA directly.
Is my software considered SaMD or SiMD?
The simplest way to distinguish SaMD from SiMD is to ask whether the software performs the medical function on its own or whether it exists to operate a hardware medical device.
SaMD performs a medical purpose independently from hardware. For example, software that analyzes medical images to help detect a disease may be SaMD.
SiMD is software that is part of a hardware medical device. For example, software that controls the delivery of insulin from an insulin pump is software in a medical device.
Let’s say you’ve determined that your product meets the definition of a medical device. You still need to consider the second half of the SaMD definitions from the IMDRF and FDA.
-
The IMDRF definition tells us that the software must perform its purposes “without being part of a hardware medical device.”
-
FDA uses almost the exact same language, stating that SaMD “is intended to be used for one or more medical purposes without being part of a hardware device.”
This narrows the scope of SaMD again. Software that is used to power hardware or drive a hardware device does not meet the criteria for SaMD.
Instead, that type of software is what’s known as SiMD, or “software in a medical device.”
Simply put, any software that helps to run a hardware medical device—say, by powering its mechanics or producing a graphical interface—is software in a medical device. Some examples include:
-
Software that controls the inflation or deflation of a blood pressure cuff
-
Software that controls the delivery of insulin on an insulin pump
-
Software used in a closed loop control of a pacemaker
These types of software are also known as “embedded software” of “firmware”, or “micro-code” so if you hear those terms, know that they indicate SiMD, not SaMD.
For a product to be classified as SaMD, the software must stand alone from any hardware as it performs the functions that categorize it as a medical device. IMDRF guidance on Possible Framework for Risk Categorization and Corresponding Considerations adds several clarifying points to the SaMD definition:
-
SaMD is a medical device and includes in-vitro diagnostic (IVD) medical device.
-
SaMD is capable of running on general purpose (non-medical purpose) computing platforms.
-
“without being part of” means software not necessary for a hardware medical device to achieve its intended medical purpose.
-
Software does not meet the definition of SaMD if its intended purpose is to drive a hardware medical device.
-
SaMD may be used in combination (e.g., as a module) with other products including medical devices.
-
SaMD may be interfaced with other medical devices, including hardware medical devices and other SaMD software, as well as general purpose software.
-
Mobile apps that meet the definition above are considered SaMD.
NOTE: While it’s important to differentiate between SaMD and SiMD, the two types of software share many of the same standards for development, such asIEC 62304,the international standard for software lifecycle processes. If your software is actually SiMD, you’ll still find many of the guidance documents and standards in this guide useful.
What are some examples of SaMD?
Theory is one thing, but it’s often easier to grasp real examples. I’ll go over a few examples of SaMD here, but you can find more in the same IMDRF guidance on risk classification mentioned above:
-
Software that allows a mobile device to view images from an MRI, ultrasound, or X-ray that are used for diagnostic purposes
-
Software that processes images to help detect breast cancer
-
Software that diagnoses a condition using the tri-axial accelerometer on a smartphone
-
Software that collects patient data in real-time that is monitored by a medical professional and used to develop treatment plans.
Common examples of SaMD
Common examples of software as a medical device include software that:
- Analyzes medical images for diagnostic support
- Uses patient data to help inform treatment decisions
- Monitors physiological data and alerts healthcare providers
- Supports clinical decision-making without directly controlling hardware
- Processes data from a mobile device, wearable, or sensor for a medical purpose
The common thread is intended use. A software product does not become SaMD simply because it is used in healthcare. It becomes SaMD when it is intended to perform a medical device function independently from a hardware medical device.
.png)
![clinical-evaluation-2-1]() How is SaMD regulated and classified around the world?
How is SaMD regulated and classified around the world?
While there are many markets for medical devices around the world, the US and the EU are by far the largest, so these are the markets we’ll focus on in this section.
The first point that I want to make is that a SaMD product is still a medical device, and is regulated as such.
For starters, you will need a quality management system (QMS). In the US, you must follow the Quality Management System Regulations (QMSR) from the FDA. Likewise, in the EU, your SaMD will be governed by the EU MDR (or EU IVDR if it is an in vitro diagnostic device). And finally, your device will still be classified according to the applicable regulations in either market.
Basically, it pays to remember that while your SaMD may be significantly different from a traditional, hardware medical device, you still need to follow the same regulations as any other medical device.
With that in mind, let’s get into some of the nuances of the SaMD regulatory landscapes in the US and EU.
How is SaMD regulated and classified in the US?
First, we have the US system for classifying SaMD. FDA classifies SaMD using the samerisk classesas it does for traditional medical devices:Class I,Class II, andClass III.
However, the FDA has also put out a guidance document on the content of premarket submissions for device software functions to help manufacturers better understand the documentation they will need to submit for devices that include software.
Note on terminology: While SaMD is the widely used term for software as a medical device, in recent guidance documents, the FDA has begun using the term “device software function” or DSF. The agency defines DSF as:
Software function that meets the device definition in section 201(h) of the FD&C Act. The term “function” is a distinct purpose of the product, which could be the intended use or a subset of the intended use of the product.
How does the FDA classify SaMD?
FDA classifies SaMD using the same device risk classes used for other medical devices: Class I, Class II, and Class III. The classification depends on the software’s intended use, indications for use, and the risk posed if the software fails or provides incorrect information.
FDA also expects manufacturers to determine the appropriate software documentation level for premarket submissions. Some device software functions require basic documentation, while higher-risk software functions may require enhanced documentation. These documentation levels do not map perfectly to device class, so each software function must be assessed based on its own risk profile.
There are two levels of documentation, enhanced and basic, which are defined by the FDA thusly:
-
Enhanced Documentation: should be provided for any premarket submission that includes device software function(s) where a failure or flaw of any device software function(s) could present a hazardous situation with a probable risk of death or serious injury, either to a patient, user of the device, or others in the environment of use. These risks should be assessed prior to implementation of risk control measures. Sponsors should consider the risks in the context of the device’s intended use (e.g., impacts to safety, treatment, and/or diagnosis), and other relevant considerations.
-
Basic Documentation: should be provided for any premarket submission that includes device software function(s) where Enhanced Documentation does not apply.
It’s the sponsor’s job to consider all the risks that are part of their device’s risk assessment (including cybersecurity risks) and individually assess their device to determine the appropriate level of documentation. However, there are some devices that the FDA recommends should have enhanced documentation, including:
-
Devices intended to test blood donations for transfusion-transmitted infections
-
Devices used to determine blood donor and recipient compatibility
-
Automated blood cell separator devices intended for collection of blood and blood components for transfusion or further manufacturing use
-
Blood establishment computer software (BECS)
The FDA also recommends enhanced documentation for:
-
Devices that are a constituent part of a combination device
-
Class III devices
Just remember, documentation levels do not correspond directly with risk class. While Class III devices will almost certainly require enhanced documentation, that does not mean Class II or Class I devices will only require basic documentation. Again, you are required to individually assess each of your devices to determine the appropriate level of documentation.
How is SaMD regulated and classified in the EU?
SaMD regulation in the EU is similar to regulation in the US, in that it does not differ from the way traditional medical devices are regulated. You will still need to comply with all the relevant requirements in the EU MDR and EU IVDR.
It’s important to note, however, that EU regulations do not use the term “software as a medical device.” Rather, they use the term “medical device software” or MDSW for short.
Fortunately, the European Commission (EC) has put out several guidance documents relevant to SaMD manufacturers.
-
MDCG 2021-24 - Guidance on classification of medical devices
-
Infographic - Is your software a medical device?
-
MDCG 2020-1 - Guidance on clinical evaluation and performance evaluation of medical device software
-
MDCG 2019-16 - Guidance on cybersecurity for medical devices
-
MDCG 2019-11 - Qualification and classification of software
I’ll get into some of these later in the guide, but for now, I’d encourage you to read or at least bookmark these resources for easy reference. You’ll find them immensely helpful, especially if you’re wondering whether your software meets the definition of medical device software in the EU.
Medical device software risk class and “Rule 11” in the EU
As with the US, there is no MDSW-specific risk classification in the EU. Medical device software uses the same risk classification as traditional medical devices: class I, class IIa, class IIb, and class III.
But the EU MDR has an outline for how you should go about determining your medical device software risk class, identified as Rule 11.
Rule 11, which can be found in Annex VIII of EU MDR, states:
Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause:
-
-
death or an irreversible deterioration of a person's state of health, in which case it is in class III; or
-
a serious deterioration of a person's state of health or a surgical intervention, in which case it is classified as class IIb.
-
Software intended to monitor physiological processes is classified as class IIa, except if it is intended for monitoring of vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb.
All other software is classified as class I.
The EC has elaborated on Rule 11 and its process for classification in MDCG 2021-24 and MDCG 2019-11. However, as you may have noticed reading Rule 11, most medical device software will be classified as at least class IIa under the rule.
If you know your device is MDSW and you want to make sure you’re complying with EU MDR requirements, using this free guidance document and gap assessment tool from Greenlight Guru is a great way to assess your compliance and determine the appropriate regulatory route for your medical device software.
SaMD categorization according to IMDRF
The IMDRF has also put out guidance on categorizing SaMD. What you’ll notice immediately when you read this document is that the IMDRF categorization has four possible categories, rather than three, and requires a table to help you identify what category your device falls under.
This table is two-dimensional, requiring the identification of both situation or condition and the significance of the information provided by the SaMD.
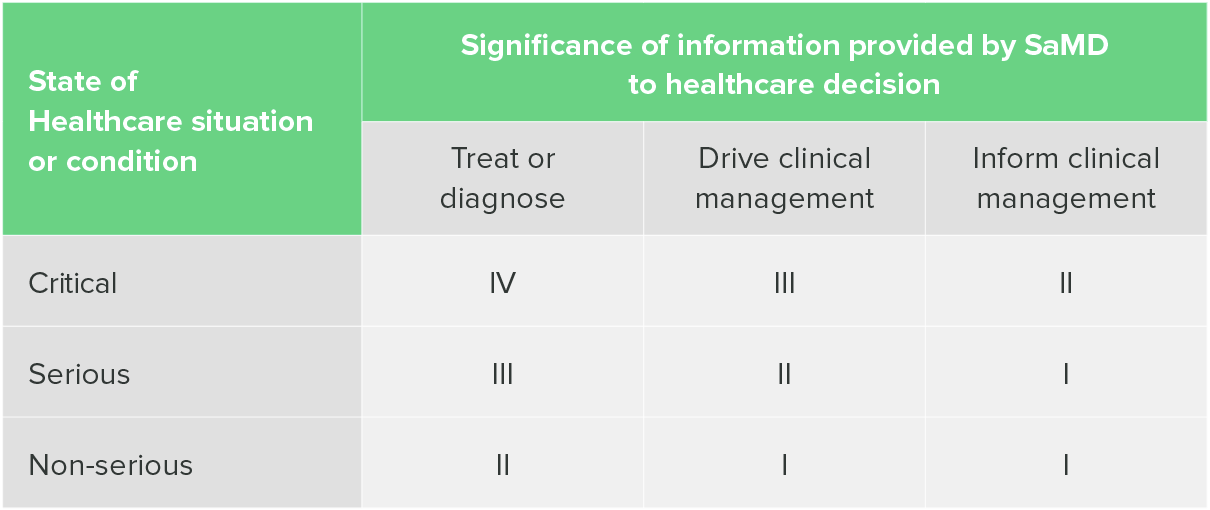
"Software as a Medical Device": Possible Framework for Risk Categorization and Corresponding Considerations, IMDRF
Currently, the IMDRF categorization is not widely used, probably because it seems to add a second, confusing layer to SaMD classification. However, the IMDRF categorization can be useful in helping to determine your risk class in the EU, and this is what most companies use it for.
One of the aforementioned EU guidance documents, MDCG 2019-11, includes a table that combines both the IMDRF categorization and EU risk classes.
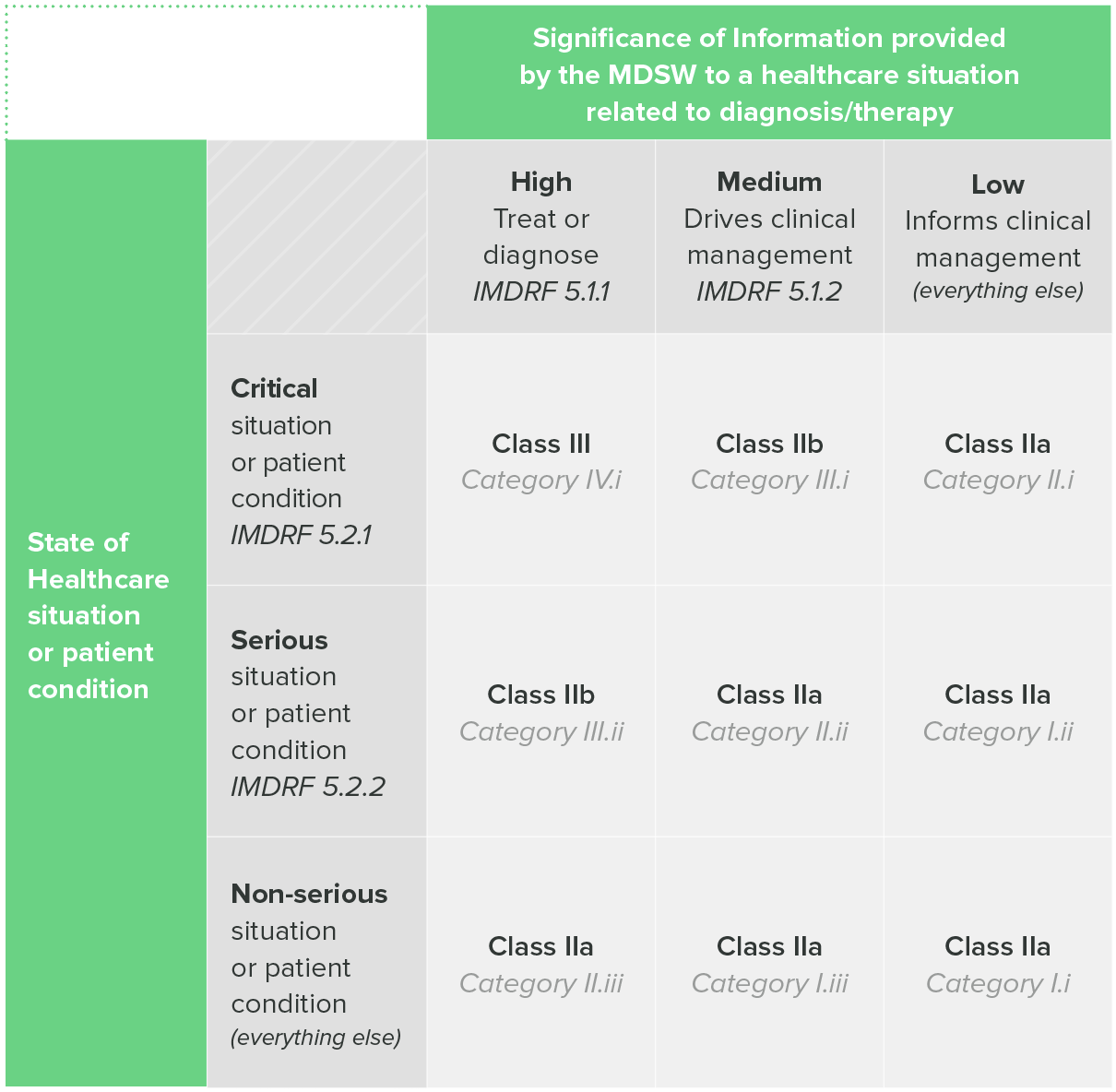
Table 1: classification Guidance on Rule II, MDCG 2019-11
So, if you intend to sell your device in the EU, the IMDRF categorization can be a useful tool for helping you determine the risk class of your SaMD.
Software safety classification according to IEC 62304
Finally, we have the software safety classification of IEC 62304, the international standard on software lifecycle processes. I’ll get into IEC 62304 in more depth later in this guide, but for now, I want to focus on its classification system.
The IEC 62304 classification system has three levels based on the severity of injury that a software failure could cause:
-
Class A - no injury
-
Class B - non-serious injury
-
Class C - serious injury or death
NOTE: Software safety classification is not a determination of how safe your software is. Instead, it is the basis for determining how rigorous your software development process must be.
The actual safety of your device depends on your design and development processes, not on a safety classification.
Something you should keep in mind is that your safety classification under IEC 62304 does not directly correspond with risk classification under US or EU regulations. Safety classification does, however, have a strong correlation with risk class.
For example, if your safety classification is class C, then there’s a good chance your device will be class III (for either US or the EU). But you could conceivably have a device that is safety class C, yet falls into a lower risk class.
The point being, the IEC 62304 classification system is about safety, but it only indicates the level of rigor you should use during software development. It is not a perfect proxy for risk classification and it does not tell you anything about the actual safety of a finished SaMD.
Does IEC 62304 safety class determine SaMD risk class?
No. IEC 62304 software safety classification does not directly determine FDA or EU medical device risk class. IEC 62304 classification is used to determine the rigor of the software development lifecycle process based on the severity of harm that could result from software failure.
Regulatory risk class and IEC 62304 software safety class are related, but they are not interchangeable. A high IEC 62304 safety class often correlates with a higher regulatory risk class, but manufacturers still need to classify the device according to the applicable regulatory framework in each market.
If you’re still with me, then give yourself a pat on the back. This is tricky stuff, but depending on where you’re planning to market your device, you may need to understand how to use every one of these classification methods.
Perhaps the most important takeaway here is that you should never assume that one classification method, such as level of concern or software safety class, will directly correspond with another.
![clinical-evaluation-3-1]() Software development for SaMD
Software development for SaMD
Start talking about software development in the medical device world, and you’re likely to hear a lot of pointed opinions.
The big reason for this is simple. A lot of regulations, like FDA’s QMSR, were written with traditional, hardware medical devices in mind. As such, they assume a very linear style of product development, where one task is accomplished before you proceed to the next. This is generally known as the waterfall methodology, and because regulations and standards like IEC 62304 are written this way, it causes a lot of angst among software developers.
Most developers today use an agile methodology, a more flexible approach based on a continuous loop of iteration. On top of that, many emerging SaMD companies don’t come from the traditional medical device world. As a result, many of these teams think it’s impossible to apply an agile methodology in medical device development based on current regulations.
So let me set the record straight right here and now. Yes, the regulations are written in a linear style that easily tracks with a waterfall approach. However, it is very much still possible to use agile product development and still maintain compliance with all the relevant regulations and standards.
For instance, when you first look at IEC 62304, you’ll notice that it has a very linear structure. But it is still possible to fulfill its requirements while using agile product development. In fact, there is another standard AAMI TIR 45 that offers guidance on the use of agile practices in the development of medical device software.
With that out of the way, let’s take a closer look at IEC 62304 and how it’s used in SaMD development.
IEC 62304 - software lifecycle requirements
IEC 62304 is a process standard, as opposed to a product standard. It will not tell you specific requirements for your product. Instead, it offers a method for structuring your processes that will result in safe and effective software, if carried out properly.
After the general requirements, IEC 62304 explains the five processes you’ll need to follow during the lifecycle of your software:
-
Software development process
-
Software maintenance process
-
Software risk management process
-
Software configuration management process
-
Software problem resolution process
Keep reading for a brief description of each of these processes.
Software development process
The software development process, according to IEC 62304, begins with software development planning and ends with the software release. Between those points, you’ll need to carry out a number of essential steps, including:
-
Software requirements analysis
-
Software architectural design
-
Software unit implementation and verification
-
Software integration and integration testing
-
Software system testing
However, once the software has been released, you’re by no means finished with your responsibilities. Remember, IEC 62304 is a software lifecycle standard, which means you'll need to maintain the software and resolve problems as they arise.
Software maintenance process
IEC 62304 lists the requirements for a software maintenance process. These requirements will look very similar to complaint handling requirements from ISO 13485 and FDA 21 CFR Part 820.
The software maintenance process outlined in IEC 62304 consists of three parts:
-
Establishing your software maintenance plan
-
Analysis of problems and modifications
-
Implementing modifications
TIP: Study your existing complaint handling process before you create a new process for software maintenance. There may already be a lot of overlap between the two, and you may not need two separate processes.
 With Greenlight Guru’s dedicated Complaint Management Software, feedback and complaints are captured in the same single system as your product’s design, management, and risk with interlinking capabilities so you can see everything impacted by customer feedback.
With Greenlight Guru’s dedicated Complaint Management Software, feedback and complaints are captured in the same single system as your product’s design, management, and risk with interlinking capabilities so you can see everything impacted by customer feedback.
Software risk management process
If you’re coming to IEC 62304 from a medical device manufacturing background, then the software risk management process it outlines should look pretty familiar to you. That’s because the risk management requirements in IEC 62304 correlate with those in ISO 14971, the international standard on the application of risk management to medical devices.
And remember, SaMD manufacturers are expected to follow ISO 14971, just like any other medical device manufacturer. If you’re unclear about anything related to risk management, then check out our Definitive Guide to ISO 14971.
You’ll quickly notice the connection between ISO 14971 requirements and those of IEC 62304, such as:
-
An analysis of software contributing to hazardous situations
-
Risk control measures
-
Verification of risk control measures
-
Risk management of software changes
Keep in mind, just because risk management gets its own section, that doesn’t mean it isn’t relevant to other processes. In fact, the software development process itself can be used as a risk control measure when it’s carried out according to IEC 62304.
Software configuration management process
Software configuration is like accounting for your software. You’re keeping track of everything you would need to recreate the software, which is foundational for traceability and release management.
The requirements laid out in IEC 62304 include:
-
Configuration identification
-
Change control
-
Configuration status accounting
Many companies use a configuration matrix to help them with configuration management. This matrix combines both the software items you want to control and when they were released in one handy table, like the one illustrated below.

The software Design and Development File (DDF)
Every piece of documentation IEC 62304 requires eventually needs to end up in the software Design and Development File (DDF). If you’re used to seeing the term software Design History File (DHF), know that the two terms (DDF and DHF) are referring to the same documentation. However, the DDF is the term that replaced the DHF when QMSR went into effect in early 2026.
Plans, requirements specifications, architecture documents, test protocols, risk records, configuration management records, and release documentation all flow into the software DDF. In aggregate, it becomes the evidence record that regulators and notified bodies examine to confirm that your software was developed in a controlled, traceable manner.
A software DDF must incorporate the full IEC 62304 development lifecycle, including the Software Requirements Specification (SRS), architectural design records, unit verification evidence, integration testing records, system test results, and all associated risk management documentation. The FDA's 2023 final guidance on the content of premarket submissions for device software functions also requires a Software Design Specification, equivalent to the detailed design documentation in IEC 62304, to be present in the DDF even when basic documentation is the applicable level. It will be reviewed during site inspections even if not submitted.
SaMD teams and Agile Development
Agile becomes a compliance problem only when sprint work is disconnected from design controls. A compliant agile process should connect user stories, requirements, risk controls, code changes, tests, defects, release notes, and approval records. The goal is not to force software teams into waterfall development. The goal is to make iterative development auditable.
Compliance in an agile environment
One of the most common questions from SaMD teams with engineering backgrounds is whether IEC 62304 requires waterfall development. The answer is no. The standard specifies what must be documented and demonstrated, not how development must be structured. Agile teams can satisfy IEC 62304 by mapping design control activities to sprint-level work and maintaining traceability within the tools they already use.
Jira, GitHub Issues, and Azure DevOps can all support the requirement-to-test traceability IEC 62304 requires, provided teams use consistent ID schemes and linking practices that create an auditable trail from a change request through implementation and verification.
Continuous integration (CI) is generally compatible with a compliant SaMD development process. Continuous deployment (CD), where code is pushed to production automatically, requires more care. Every release to end users must be documented as a product release, with updated documentation including instructions for use, release notes, and a software release record. If CI/CD pipelines are configured to trigger documentation updates and traceability checks automatically, compliant continuous deployment is achievable. If they aren't, the documentation will lag behind the software, and the DDF will have gaps.
The AAMI TIR 45 guidance, referenced earlier in this chapter, provides the framework for mapping agile sprint activities to IEC 62304 process requirements. Teams that want to use agile development while maintaining compliance should start there.
Software problem resolution process
The term “software problem resolution process” is a bit of a misnomer, because it implies that you need one problem resolution process. In reality, you will need several different problem resolution processes because you’ll encounter different types of problems throughout the software lifecycle.
IEC 62304 does lay out a general outline for a problem resolution process:
-
Prepare problem reports
-
Investigate the problems
-
Advise relevant parties
-
Use change control process
-
Maintain records
-
Analyze problems for trends
-
Verify software problem resolution
-
Test documentation contents
As a rule of thumb, you’ll likely encounter fewer problems over time as your product matures. However, the severity of the problems you encounter later on are likely to be more severe—and will require a more rigorous process for resolution.
Looking ahead: IEC 62304 Edition 2
IEC 62304:2006+A1:2015 has served as the foundational standard for medical device software development for nearly two decades. A second edition is currently in the final stages of the standardization process, with publication targeted for August 2026. That said, this revision has been in development for some time, and the previous attempt at updating the standard stalled before completion. Formal regulatory adoption by FDA and EU bodies will follow publication on its own timeline, and could take two to three years after publication.
However, even though formal regulatory adoption is likely a few years down the road, teams working on SaMD should be prepared for the changes that are coming in the new edition of IEC 62304. That’s why we’ve included a roundup of some of the most important changes to be aware of below.
Once the final standard is published, we’ll update this guide accordingly.
Simplified classification: two rigor levels replace three safety classes
IEC 62304 Edition 2 will replace the current three-class safety classification system with two Software Process Rigor Levels. Level I will correspond roughly to the current Class A, covering software where no injury or damage to health is possible. Level II will replace both Class B and Class C, capturing any software where a failure could cause injury of any kind.
For teams currently operating at Class B, this is the most consequential change in the draft. Class B teams will fall under Level II requirements, which align more closely with the current Class C standard. A gap analysis against Level II requirements is worth beginning before Edition 2 is formally adopted. Class C teams will largely find their existing processes already compatible with Level II expectations.
This change will also align IEC 62304's classification logic more closely with FDA's Basic/Enhanced documentation framework, a useful convergence for teams managing simultaneous FDA and EU submissions.
Expanded scope: all health software
Edition 1 applies specifically to medical device software. Edition 2 will expand the scope to cover all health software, including clinical decision support tools, hospital IT systems, and wellness-to-clinical transition applications that sit in the regulatory gray zone between consumer and medical device.
For SaMD manufacturers, this does not reduce obligations. Process rigor still applies, and ISO 13485 and ISO 14971 remain required elements of a regulatory submission. The practical effect is that IEC 62304 will become the reference framework for a broader category of software that may touch a product ecosystem.
AI/ML lifecycle requirements
For the first time, IEC 62304 Edition 2 will include explicit requirements for AI and machine learning development. Manufacturers building AI-enabled SaMD will need to define phases of the AI lifecycle, covering data collection, training, validation, deployment, and post-market monitoring, and treat each phase as part of software development planning. The draft also includes an informative annex (Annex E) that will provide guidance on managing adaptive and continuously learning systems, maintaining traceability when design is partially determined by training data rather than explicit specification, and approaching explainability in a safety context.
A clearer line between development and maintenance
Edition 2 will explicitly define a distinction that Edition 1 implied but did not draw cleanly. "Development" will cover new software and new features. "Maintenance" will permit a smaller process for implementing rapid changes in response to urgent problems. For teams managing frequent software releases, this distinction has direct workflow implications: a feature change triggers the development process, while an urgent patch does not.
QMS requirements move out of the standard
The general quality management system requirements currently in Clause 4.1 will be removed from IEC 62304 Edition 2, shifting that responsibility to a manufacturer's broader ISO 13485-based QMS. IEC 62304 processes will need to be nested clearly within QMS documentation rather than treated as a parallel system.
Planning ahead
Edition 2 will not be immediately required after publication. EU harmonization and FDA recognition both follow their own timelines, typically two to three years after a standard's release. Teams building new products now should identify which rigor level their software components would fall under in the new model and flag any Class B components for a future gap analysis.
Software validation for SaMD development
According to the FDA’s Quality System Regulations,
When computers or automated data processing systems are used as part of production or the quality system, the [device] manufacturer shall validate computer software for its intended use according to an established protocol.
For those coming from a software development background, without much insight into the medical device industry, the requirement to validate software that’s used to build other software may be an unexpected speed bump. However, this type of validation is essential to producing safe medical devices, whether they’re hardware or software.
How much validation is required? Well, in FDA’s guidance on the General Principles of Software Validation, it states, “The level of validation effort should be commensurate with the risk posed by the automated operation.”
For instance, if the software application that you’re using for testing doesn’t work correctly and gives you a false pass, or doesn’t test everything it should have, then you’re looking at a serious issue that could affect patient health and safety.
Additionally, in February 2026, the FDA released their final guidance document on Computer Software Assurance for Production and Quality System Software. This guidance lays out the concept of computer software assurance (CSA), which is the risk-based approach to testing software.
As FDA states:
This guidance describes a risk-based approach to establish confidence in the automation used for production or quality management systems, identify where additional rigor may be appropriate, and various methods and testing activities that may be applied to establish computer software assurance. FDA’s goal is to help manufacturers produce high quality medical devices while complying with the Quality Management System regulation, 21 CFR Part 820.
The goal of the CSA guidance is to help medical device companies reduce the overall burden of software validation while allocating resources to ensure that high-risk software is meeting its intended uses.
Just remember that all of your validation must be done in accordance with a documented protocol. And the results of that validation must be documented, as well.
Ultimately, the decision about whether or not to validate a software tool comes down to you. Keep in mind, however, that whatever your decision, you will be expected to justify it.
SOUP and SaMD Manufacturer Responsibilities
SOUP matters because most SaMD products rely on third-party software components that the manufacturer did not develop. These may include open-source libraries, operating systems, frameworks, database tools, cloud components, or other off-the-shelf software.
Even though the manufacturer did not create those components, the manufacturer is still responsible for the safety and performance of the finished medical device. That means each SOUP or OTS component should be identified, assessed for potential hazards, verified for its intended use, monitored for vulnerabilities, and controlled through configuration management.
SOUP and OTS component tracking
Modern software is almost never built from scratch. Virtually every SaMD product includes third-party libraries, open-source frameworks, operating systems, communication stacks, database engines, or hardware drivers. IEC 62304 has a specific term for these components: Software of Unknown Provenance, or SOUP.
The standard defines SOUP as "a software item that is already developed and generally available and that has not been developed for the purpose of being incorporated into the medical device, or software item previously developed for which adequate records of the development processes are not available." FDA uses the term Off-the-Shelf Software (OTS) to describe largely the same category. In regulatory submissions, the terms are effectively interchangeable. The assessment requirements under both frameworks are the same.
The core issue with SOUP is loss of lifecycle control. Manufacturers who use third-party components retain full responsibility for device safety while giving up visibility into how those components were developed, how bugs are reported, and how vulnerabilities will be patched. IEC 62304 addresses this by requiring manufacturers to formally document each SOUP item, assess the hazards it could introduce, verify it meets the requirements for its intended use, and track it under configuration management.
The required documentation varies by software safety class. Class A devices require basic identification and anomaly review. Class B and C devices require the full assessment: a written definition of what the SOUP item must do, a documented hazard analysis for failure modes introduced by the component, evidence of testing, and an ongoing plan for monitoring vulnerabilities through the device's postmarket lifecycle.
For SaMD teams, SOUP management has a direct relationship with the SBOM discussed in the cybersecurity chapter. An SBOM is essentially the inventory that makes SOUP tracking scalable. It documents every component and its version, dependencies, and available provenance information. Without a complete, maintained SBOM, keeping the SOUP documentation current as dependencies are updated becomes an unmanageable manual process.
![clinical-evaluation-4-1]() Cybersecurity and SaMD
Cybersecurity and SaMD
Safety is always paramount when it comes to medical devices. And creating safe SaMD means considering an extra element: cybersecurity.
Whether you’re coming from the software development world or the medical device industry, the need for cybersecurity measures should be clear by now. There have been a number of high-profile attacks in the healthcare industry in the past decade, and new vulnerabilities are being discovered all the time. In fact, healthcare is regularly cited as one of the most at-risk industries for cyberattacks.
All of this means that device makers can no longer afford to put cybersecurity on the back burner or try to add it into a finished device. The threats are real, and it’s critical that you take them seriously to protect the safety of your device users.
Software bill of materials (SBOM) for software as a medical device
On May 12, 2021, the Biden administration issued the Executive Order on Improving the Nation’s Cybersecurity. One important aspect of this executive order was its focus on a software bill of materials (SBOM). Congress then passed the PATCH Act in 2022 (which came into effect on March 29, 2023) codifying the need for an SBOM and requiring manufacturers to address postmarket cybersecurity vulnerabilities.
An SBOM is a nested inventory of all third party software that exists within SaMD or SiMD. A software bill of materials is crucial to the safety and security of your product, because it provides a list of ingredients for your device.
If a vulnerability is found in some widely used software component, you can quickly check whether that component is in your list of ingredients. If it is, you’ll know which products are affected and can quickly alert providers and work to fix the issue.
The executive order directed the National Telecommunications and Information Agency (NTIA) to create a list of the minimum elements that are required for an SBOM, which the NTIA has since published. The minimum elements include:
-
Data Fields - Document baseline information about each component that should be tracked: Supplier, Component Name, Version of the Component, Other Unique Identifiers, Dependency Relationship, Author of SBOM Data, and Timestamp.
-
Automation Support - Support automation, including via automatic generation and machine-readability to allow for scaling across the software ecosystem. Data formats used to generate and consume SBOMs include SPDX, CycloneDX, and SWID tags.
-
Practices and Processes - Define the operations of SBOM requests, generation and use including: Frequency, Depth, Known Unknowns, Distribution and Delivery, Access Control, and Accommodation of Mistakes.
Now, you may be looking at these minimum elements and thinking that this looks like a lot of work to put together. And you’re not wrong—it’s true that compiling an SBOM on your own and monitoring every component for new vulnerabilities is a heavy lift. This is why I’d recommend using an automated SBOM tool that will compile all of this for you and proactively monitor the elements in your SBOM for new vulnerabilities.
There are a number of tools out there for you to use, including the SBOM Analysis and Vulnerability Management Tool from our partners at MedCrypt. I’d encourage you to explore your options before building your SBOM manually.
Threat modeling and cybersecurity risk management
The most recent cybersecurity guidance documents (referenced at the end of this chapter) are clear that cybersecurity risk must be assessed through threat modeling, rather than treated as an extension of the safety risk management process already required by ISO 14971.
Safety risk management, conducted under ISO 14971, uses a probabilistic model that evaluates the likelihood of harm and its severity. FDA's 2025 final cybersecurity guidance is explicit that cybersecurity risk requires a nonprobabilistic model instead, one focused on exploitability and potential impact rather than probability. A traditional Failure Mode and Effects Analysis (FMEA) alone is not sufficient for cybersecurity. A threat model fills the gap that FMEA leaves open.
Threat modeling for a medical device follows a structured process. The starting point is defining the system boundary and creating an accurate view of the security architecture: what are the components of the device, where does data enter and leave, what are the interfaces, and what assumptions are being made about the operating environment? From there, threats are identified systematically, often using the STRIDE methodology (Spoofing, Tampering, Repudiation, Information disclosure, Denial of service, Elevation of privilege), which analyzes each component and connection for the specific threat categories that apply.
FDA's guidance states that the threat model must identify device system risks and mitigations, inform the pre- and post-mitigation risk assessment that is part of the cybersecurity risk analysis, document assumptions about the operating environment, and capture risks introduced through the supply chain, manufacturing, deployment, interoperability, maintenance, and decommissioning. These are categories that a traditional safety FMEA will often miss entirely.
The relationship between threat modeling and FMEA is parallel, not hierarchical. Both are structured analyses of a system looking for failure modes, and the resulting records often inform each other. A cybersecurity threat that triggers device failure, for example, may surface as a hazardous situation in the ISO 14971 risk file. A safety-critical software component identified through FMEA should receive additional scrutiny in the threat model.
FDA's guidance recommends that threat modeling begin once the system architecture is reasonably well defined, and that it be updated throughout the product lifecycle rather than treated as a one-time premarket activity. For SaMD, this is particularly relevant, as software changes, new integrations, and evolving threat environments all require the threat model to be revisited.
The MITRE Playbook for Threat Modeling Medical Devices, referenced in the guidance list below, is FDA's recommended starting point for teams building a threat modeling process for the first time.
Cybersecurity regulations, guidance, and resources
Cybersecurity is a relatively new and ever-evolving field for regulators and medical device professionals alike. However, regulatory bodies are catching up, publishing guidance documents and resources that every SaMD professional should read and understand.
I’ve compiled a short list of them here and it’s a good place to start if you’re wondering what regulatory bodies will expect from you and your product regarding cybersecurity:
-
Playbook for Threat Modeling Medical Devices. Threat modeling is one of the best methods for strengthening the security and safety of your SaMD. That’s why FDA has developed this threat modeling playbook. In it you’ll find resources for developing and adapting your company’s threat modeling practices.
-
Cybersecurity for Networked Medical Devices Containing Off-the-Shelf (OTS) Software. This is the FDA guidance on the use of off-the-shelf (mass marketed) software in medical devices. A vulnerability in this type of software may pose a risk to the operation of medical devices and generally requires ongoing maintenance throughout the product lifecycle. This guidance clarifies how regulations like QMSR apply to cybersecurity maintenance activities.
-
Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions This document provides FDA’s recommendations to industry regarding cybersecurity device design, labeling, and the documentation that FDA recommends be included in premarket submissions for devices with cybersecurity risk
-
Postmarket Management of Cybersecurity in Medical Devices. This FDA guidance document provides the agency’s recommendations for managing postmarket cybersecurity vulnerabilities in medical devices. It offers specific recommendations and encourages a total product lifecycle approach to cybersecurity.
-
MDCG 2019-16 - Guidance on Cybersecurity for Medical Devices. This guidance document is meant to help manufacturers fulfill the requirements of Annex I of both EU MDR and EU IVDR, with regard to cybersecurity. It also includes the expectations from other stakeholders, such as integrators, economic operators, and users.
-
IMDRF Principles and Practices for Medical Device Cybersecurity. This guidance was issued to offer best practices and general principles for medical device cybersecurity. The IMDRF’s stated goal for the guidance is to “facilitate international regulatory convergence on medical device cybersecurity,” and it offers recommendations for both device manufacturers and external stakeholders, such as providers.
For an even deeper dive, check out the cybersecurity page from FDA, which contains a lengthy list of news and updates, white papers and reports, guidance documents, known vulnerabilities, and other resources on cybersecurity.
If there’s one thing I can impress upon you about cybersecurity, it’s that you need to start thinking about it early on. The expectation is that you’re addressing cybersecurity throughout the design process, rather than treating it as an “extra” that’s tacked on once your product is finished.
The sooner you start thinking about cybersecurity for your SaMD, the easier it will be to meet regulatory expectations. Most importantly, however, baking cybersecurity into the design of your device will result in a safer, more secure product for the patients and providers using it.
![clinical-evaluation-5-1]() Postmarket requirements for SaMD
Postmarket requirements for SaMD
It bears repeating that while there are certainly nuances and added issues to deal with regarding SaMD (like cybersecurity), software as a medical device is still subject to the same regulations as hardware medical devices.
This means all of the postmarket requirements from regulations like FDA’s QMSR, EU MDR or IVDR still very much apply to your SaMD. Additionally, if you’ve been following IEC 62304 for software development, your software maintenance process and software problem resolution process will help you fulfill some of these postmarket requirements.
For a more comprehensive understanding of these regulations, check out some of the postmarket-related guides, podcasts, and webinars from our library of free medical device resources.
With that said, let’s talk about some of the features inherent in SaMD that require a different approach in the postmarket stage of a medical device’s lifecycle.
When does a SaMD require a new submission?
Making a change to a medical device that is on the market will always require scrutiny. At the very least, you’ll need to document the change you’ve made. But if a change is significant enough, it may require you to resubmit documentation you needed to get the device on the market in the first place.
When your product is software, deciding whether you need a new submission is even more complicated. So, let’s take a look at how you’ll make that decision.
Making changes to SaMD in the US
Software as a medical device is similar to traditional medical devices in that if you want to makes changes to your device, you have two options:
-
Notify FDA of the change via a new 510(k), a special 510(k), or PMA supplement
-
Document the change internally via a letter-to-file
The choice you make depends on the significance of the change you’re making and whether it affects the safety, efficacy, or performance of the device. For a hardware medical device, it’s usually easier to know whether your change is significant enough to warrant notifying FDA. But with software, the decision can be a little trickier.
Fortunately, FDA has released a guidance on Deciding When to Submit a 510(k) for a Software Change to an Existing Device to help SaMD manufacturers. As with all the guidance documents listed in this guide, I encourage you to read the entire thing. However, this flowchart will give you an idea of the questions you should ask during the decision-making process.
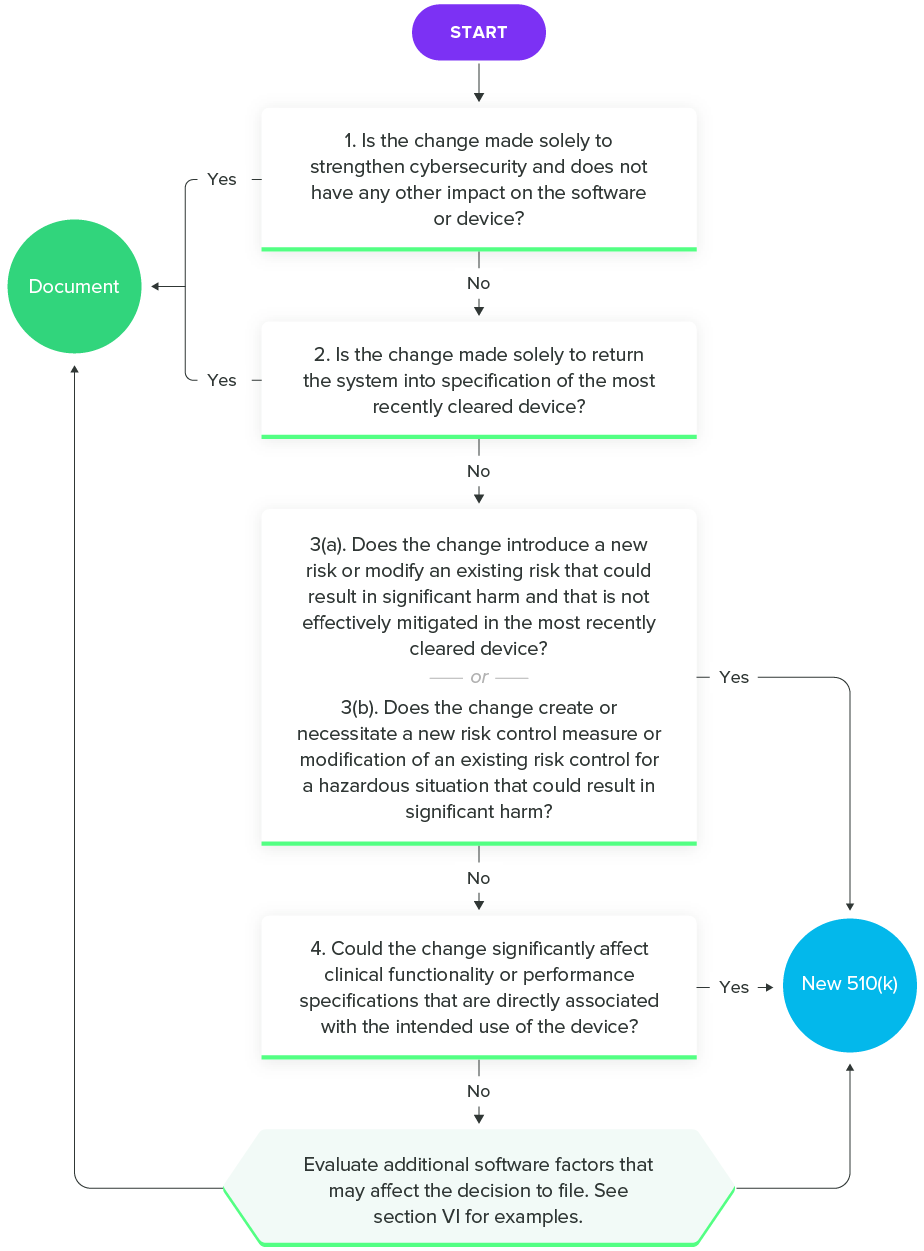
Deciding When to Submit a 510(k) for a Software Change to an Existing Device, FDA
Making changes to SaMD in the EU
The requirements for a notification of a change in your device in the EU are similar. Annex X of EU MDR states:
The applicant shall inform the notified body which issued the EU type-examination certificate of any planned change to the approved type or of its intended purpose and conditions of use.
Changes to the approved device including limitations of its intended purpose and conditions of use shall require approval from the notified body which issued the EU type-examination certificate where such changes may affect conformity with the general safety and performance requirements or with the conditions prescribed for use of the product. The notified body shall examine the planned changes, notify the manufacturer of its decision and provide him with a supplement to the EU type-examination report. The approval of any change to the approved type shall take the form of a supplement to the EU type-examination certificate
Changes to the intended purpose and conditions of use of the approved device, with the exception of limitations of the intended purpose and conditions of use, shall necessitate a new application for a conformity assessment.
Although the MDR requirements are not exactly the same as the FDA guidelines for making changes to SaMD, you should be able to notice a similar theme running through both: modifications that change the intended use of the device require a new submission to FDA or application to your notified body.
Artificial intelligence and machine learning in SaMD
There’s one more wrinkle in the decision-making process for a new submission: artificial intelligence (AI).
AI is one of the fastest growing technologies being applied to medical devices, and that includes machine learning (ML), a subset of AI that has exploded in use across many industries in recent years.
Machine learning refers to an algorithm that has the ability to change or improve its outputs as it “learns” from an increasing amount of inputs. The more data an ML algorithm receives, the more accurate its results can become.
This technology offers incredible potential for diagnostics and many other medical devices, but it does pose a dilemma. When we’re talking about SaMD, the change in the algorithm that allows it to improve is technically a change in the device itself.
So, how do you know if that change has reached the level of requiring a new submission for your SaMD?
In 2019, FDA released a Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) to begin addressing this issue. While I highly recommend reading the full document, there are a couple things I want to point out here.
First, FDA still expects manufacturers to refer to and use the guidance document, Deciding When to Submit a 510(k) for a Software Change to an Existing Device, which we discussed earlier in this section.
However, FDA has also created guidance for what it calls a Predetermined Change Control Plan (PCCP). Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence-Enabled Device Software Functions is a mouthful, but it includes some important information for companies with devices that use AI/ML.
The PCCP is a document that a manufacturer includes along with their marketing submission. The PCCP establishes the parameters of any anticipated modifications to your device's algorithm before it's on the market. It describes the modifications that will be made to the device and how they will be assessed.
Though the PCCP can become complex depending on the device, its essential parts are fairly straightforward. You need to be able to describe the modifications that will occur, how you’ll implement them, and an assessment of their risk and benefits. Here’s what the FDA expects in a PCCP:
-
Description of Modifications: This section details the planned changes, such as retraining algorithms or incorporating new datasets, and how they support device improvements.
-
Modification Protocol: This includes specific methodologies for:
-
Data management practices.
-
Retraining algorithms.
-
Performance evaluation.
-
Update procedures to ensure consistent safety and effectiveness.
-
-
Impact Assessment: Manufacturers must evaluate the risks and benefits of proposed changes and outline risk mitigation strategies.
The PCCP is meant to help manufacturers avoid a situation where they are constantly re-submitting to FDA as their algorithm learns. The agency is saying that MedTech companies may instead use a PCCP to lay out the boundaries of any changes to the algorithm in advance. By obtaining FDA authorization for this plan as part of the initial marketing submission, manufacturers can implement approved modifications without needing additional submissions.
The AI Act and high-risk AI systems
The EU's Artificial Intelligence Act (AIA) came into force in August 2024 with goal of fostering responsible AI development and deployment in the EU. The act categorizes AI in medical devices as "high-risk," meaning it must comply with specific requirements within the act. While we won't cover the AIA in full here, it's worth noting that the MDCG has put out a guidance document that covers the interplay between the AIA and EU MDR and EU IVDR.
Section V of this guidance document covers Substantial Modifications/Significant Change to high-risk AI. The guidance states:
In accordance with Article 43(4) of the AIA, high-risk MDAIs that have been subject to a conformity assessment procedure shall undergo a new conformity assessment procedure in the event of a substantial modification, regardless of whether the modified system is intended to be further distributed or continues to be used by the current deployer.
However, the guidance goes on to say that:
In addition to MDR/IVDR requirements for the implementation of procedures to manage updates and modifications to MDAIs, high-risk MDAIs that continue to learn after being placed on the market or put into service have the possibility of having predetermined changes plan checked at the time of the conformity assessment (Article 43(4) AIA). Changes to high-risk MDAIs that have been pre-determined by the manufacturer and assessed at the moment of the initial conformity assessment and are part of the information contained in the technical documentation referred to in AIA Annex IV point 2(f) shall therefore not constitute a substantial modification.
So, much like the FDA's PCCP guidance, the MDCG guidance is saying that if a change was pre-determined (and assessed during the initial conformity assessment) and is recorded in the device's technical documentation, then it would not constitute a substantial modification that requires a new conformity assessment.
Finally, in addition to the EU's AIA and FDA's guidance documents, FDA, Health Canada, and the United Kingdom’s Medicines and Healthcare Products Regulatory Agency have come together to issue guiding principles for Good Machine Learning Practice for Medical Device Development. This resource is short and to the point, and it’s essential reading for anyone building a product that includes ML.
This is still a developing field, but these principles should help guide manufacturers interested in using AI/ML technologies with a SaMD.
![clinical-evaluation-5 (2)]() Final thoughts on software as a medical device
Final thoughts on software as a medical device
If you’ve made it to this point of the guide (even if you’ve just skimmed the headers), I think you’ll agree that the world of SaMD is both complex and evolving.
There are still gray areas within regulations and guidances for SaMD that will need to be addressed in the coming years. There is still much work to be done around issues like cybersecurity. And there’s a steep learning curve for software developers entering the medical device world, and vice-versa.
But there’s also immense opportunity and excitement in this space. The last thing I want is for you to finish this guide and be put off by the work that goes into building safe and effective software as a medical device. With the right tools, and the best expert advice on hand, there’s no reason why your company can’t create high-quality SaMD that improves the quality of life for millions of patients.
At Greenlight Guru, we understand the unique dynamics of software development in the medical device industry. Our QMS acts as a bridge between your development work and regulatory requirements, providing the necessary guardrails to ensure compliance without stifling innovation. Learn what Greenlight Guru can do for you.Get your free demo ➔
The Ultimate Guide to Software as a Medical Device (SaMD)

%20Ultimate%20Guide%20to%20SaMD.png?width=5100&name=(cover)%20Ultimate%20Guide%20to%20SaMD.png)
%20Ultimate%20Guide%20to%20SaMD.png?width=250&height=324&name=(cover)%20Ultimate%20Guide%20to%20SaMD.png)



