
As a medical device professional, you are well aware of how much time and effort goes into getting your product to market. So, with so much care put in every part of the device, shouldn’t you be taking the same sharp eye to detail when it comes to the language you use in marketing it?
You have likely heard the terms Clearance, Approval, and Granted used many times throughout the medical device industry. You’ve likely heard them used interchangeably. You’ve likely even heard them used incorrectly.
But the terminology you use does in fact matter. FDA has provided definitive guidance that the terms Clearance, Approval, and Granted each have their own unique meaning for a medical device regulatory submission route.
In worst cases, using it incorrectly can result in serious legal consequences for your business and the device you worked so hard to bring to market.
It’s important that companies know the differences between the three terms and understand when it is appropriate to use them. Let’s dig in and look at the distinctions of FDA Clearance vs. Approval vs. Granted:
What is the difference between FDA Clearance, Approval and Granted for medical devices?
The terms FDA Cleared, Approved, and Granted each relate to a distinct process in one of the FDA regulated pathways to bring a medical device to market.
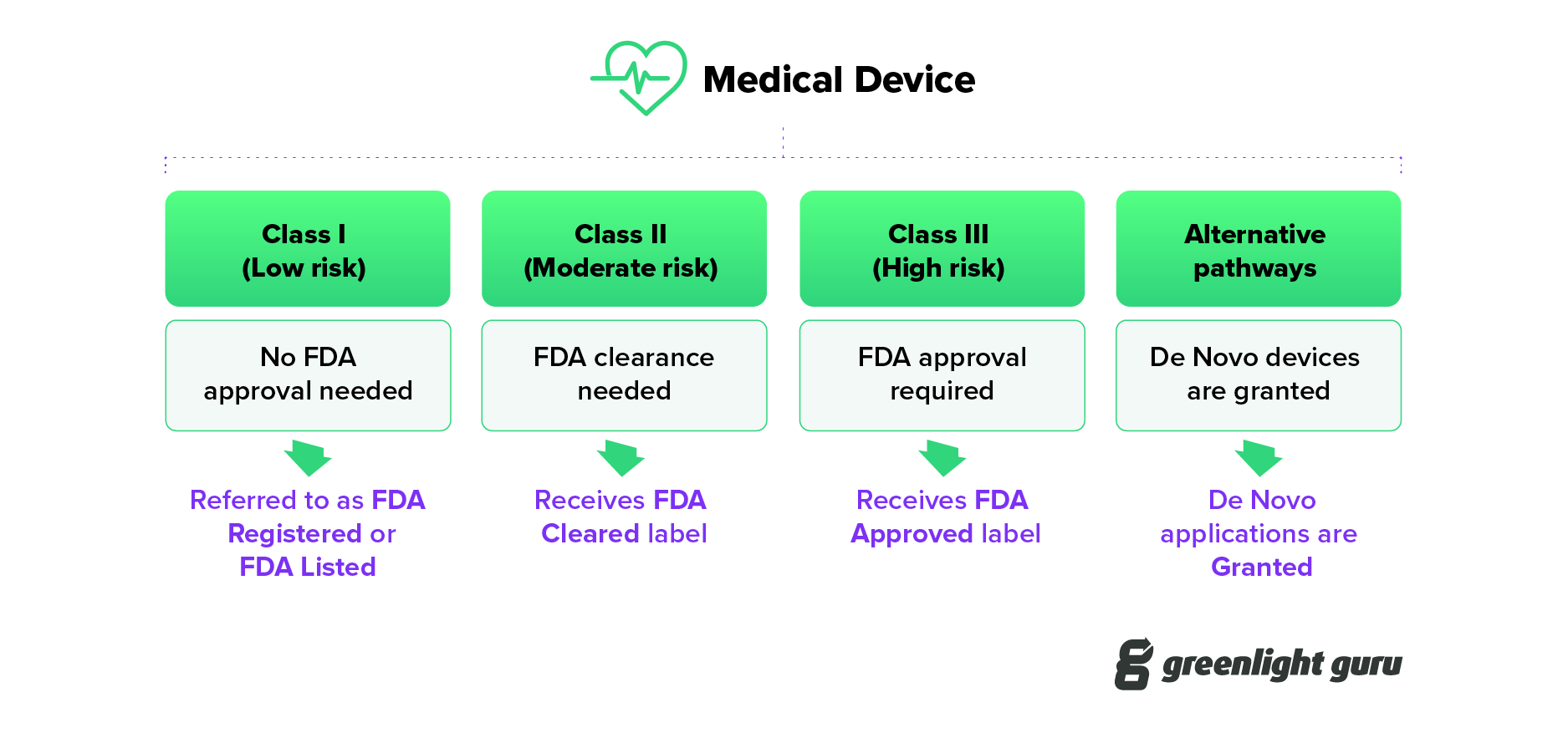
The language used by FDA changes based on the levels of associated risk. And with those changes come different definitions for regulatory requirements.
- Clearance: What does "FDA cleared" mean? When a medical device is cleared, this means it has undergone a 510(k) submission, which FDA has reviewed and provided clearance.
- Approval: For Class III medical devices to be legally marketed they must undergo a rigorous review and approval process. Following a successful submission of a premarket approval (PMA) or a Humanitarian Device Exemption (HDE), the device is given Approval by FDA.
- Granted: Medical devices using the De Novo pathway must be Granted by FDA before they can be legally marketed in the United States. This is a relatively new term in the FDA lexicon.
First FDA registered, then FDA cleared, approved or granted
In accordance with the legislation 21 CFR, all devices must be registered with the FDA. However, manufacturers must first determine device classification, which is grouped into three groups based on the overall risk level:
-
Class I for low-risk devices
-
Class II for devices with moderate risk
-
Class III for devices with the highest risk
Determining your device classification will help manufacturers determine the ideal pathway for a regulatory submission. Only then can the device be registered with the agency, effectively beginning the FDA review.
Now, I’m sure this process is nothing new to MedTech pros. But still, our industry still seems to have a fundamental misunderstanding of how these three words — clearance, approval, and granted — are all purposeful.
FDA uses them to signify something specific about that device’s risk level and the manufacturer’s ability to demonstrate safety and effectiveness before its release as a legally marketed device.
What’s the difference between 510(k) submissions vs premarket notification applications?
Looking back over the definitions of FDA cleared vs approved vs granted, we can see the clear distinction between these terms. For Class I and Class II, the FDA doesn’t give “approval,” it just gives clearance.
Greenlight Guru founder Jon Speer weighed in on the topic during an interview with The Verge, “Class I and Class II products are lower-risk products, a classic Class I example is something like a tongue depressor — and it’s much easier to get clearance than approval.”
Take the Apple Watch for example, which has garnered a lot of media attention for achieving FDA clearance as a Class II medical device. For Class II and Class I, the FDA doesn’t give “approval,” it just gives clearance.
The misuse in terminology is also commonly seen in the media where these three terms are interchangeably used and imprinted somewhere in our memories to confuse us later. I’ll see press releases, or blog posts written by large, well-known medical device companies saying their 510(k) was just “approved.”
The notion of a 510(k) premarket notification is one of the more common routes for getting a product to market. Medical device companies can use this pathway if they are able to prove substantial equivalence with a predicate device. The FDA reviewer is reviewing the information you provide to “clear” your devices as similar to the predicate.
The PMA application, which typically involves Class III devices, is much more rigorous than the 510(k) process. The device manufacturers use clinical evidence to prove a device is safe and effective, and that the benefits of the product outweigh the associated risk. This allows the FDA to put their stamp of “approval” on the device.
What happens when you get FDA clearance vs approval wrong?
Using the wrong terminology may seem like a minor clerical issue. However, it’s important to focus on why this kind of language is regulated in the first place — to ensure the safety and protection of patients, providers, and all end-users of medical devices.
When statements on product labeling or marketing materials misrepresent these terms, regardless of whether it’s a mistake or intentional, the result could be quite serious.
With that in mind, it should come as no surprise that using the wrong terminology around FDA cleared vs approved vs granted can also cause issues for your company — with both regulatory bodies and customers, alike.
How does FDA terminology impact regulatory activities?
It’s no secret that Class II and Class III products are subject to various types of FDA inspections. FDA inspectors can be some of the most detail-oriented people you will ever meet. They are going to examine your documentation very closely, so you want to make sure you get the simple things, such as terminology, correct.
Let’s say an inspector shows up at your door expecting to review a Class II device and sees that your website labels it as “approved.” That will be an immediate red flag to that inspector. If that happens, I can guarantee that the inspector will be more likely to dig deeper and deeper into your other processes, as well.
Perception is everything during an inspection. You do not want to start off on the wrong foot due to a simple case of mistaken terminology. Don’t forget that inspectors are likely to do a lot of research on your company. They will likely look at your website and any other marketing collateral they can find.
A lot of people fail to realize that much of that information is considered part of the device labeling. You want to make sure all those things get properly reviewed under your quality management system to prevent any simple mistakes from happening.
How does using FDA cleared vs approved vs granted impact your customers?
Inspections are often the main concern of quality and regulatory professionals, but there can be legal ramifications to using the wrong terminology when it comes to naming FDA cleared vs approved vs granted devices, as well.
One thing commonly professed by product liability attorneys is that if opposing counsel can object to any aspect of testimony by proving that something which was said is incorrect, the entire testimony can be thrown out. In other words, if it ever comes down to a court case, you could find yourself on the wrong side of things if incorrect terminology is used.
Consumers are also becoming increasingly more educated about medical devices due to the growing trend of wearable technology. These end-users are continually searching the internet and social media for information about these products. So much so that they may even make their buying decisions based on the terminology that is used with the device.
A simple mistake in terminology can cause some of the most powerful marketing campaigns around to have a detrimental, opposite effect on potential patients and clinicians.
Who do the correct terms matter to? Clearly quality professionals, regulatory bodies such as FDA, members of the judicial system, and even consumers. If a medical device company can’t get basic terminology right, what else might they be getting wrong?
An all-in-one MedTech solution for your product, regulatory & quality needs
Terminology is very important. You need to know your Clearances from your Approvals and Granteds.
Using these terms incorrectly can lead to a very difficult inspection, create serious legal ramifications for your company, and can even cause you to lose market share.
Launching a medical device is the culmination of a lot of hard work, time, and money. Make sure you and your colleagues understand the subtle differences in terminology. It is a simple way to prevent any self inflicted wounds that can have unfortunate consequences for your business.
For years, Greenlight Guru has brought deep expertise to medical device companies around the world through its leading QMS software and Guru Edge.
Our end-to-end platform integrates cross-functional teams, processes, and data throughout the product lifecycle, giving visibility and traceability beyond compliance requirements to proactively overcome execution gaps and accelerate success.
The Guru Edge consists of comprehensive, industry-guiding services, tools, and educational resources that accelerate your success. Greenlight Guru gives you an edge in your medical device development processes so that you can deliver
life-changing MedTech. Together, we can enhance your team with an extension of our dedicated industry experts who will help you efficiently navigate medical device regulations, reduce risk, and stay on track to accomplish your goals.
Want to see it in action? Get your free, personalized demo of Greenlight Guru today →
BONUS RESOURCE:
Content Toolkit for Medical Device Startups
Content Toolkit for Medical Device Startups






