







For many MedTech companies, especially those that manufacture high-risk devices, clinical trials are a crucial part of getting medical devices to market and keeping them there. In fact, clinical activities, both pre-market and post-market, were some of the top business objectives that respondents to our 2024 State of the MedTech Industry Benchmark Report told us they were focused on this year.

And while the clinical trial itself is extremely important, the way that a sponsor collects and manages the data the trial generates is often just as crucial. Because clinical data management is more than just rows and columns—it’s the overall process of collecting and managing research data to produce high-quality information and reliable results in compliance with local regulations and good clinical practice.
Without good clinical data management, the data from clinical trials may not accurately convey the outcomes of studies and/or the data may not be usable in the eyes of regulatory agencies.
In this guide, we’re going to cover the entire spectrum of clinical data management: how clinical data is collected, best practices for managing it in a compliant manner— as well as pitfalls to avoid—and the tools and resources available to help you in your clinical operations.
Before we get into the full guide, here are three quick answers for teams evaluating clinical data management software for medical device studies.
What is clinical data management software?
Clinical data management software is a controlled system for collecting, validating, reviewing, cleaning, locking, exporting, and retaining clinical study data. In medical device studies, it typically supports eCRFs, ePRO/eCOA, adverse events, PMCF surveys, audit trails, role-based access, electronic signatures, Discrepancy Management, Source Document Verification, and database lock workflows so sponsors can generate reliable clinical evidence for regulatory submissions and post-market requirements.
What is the difference between CDMS and EDC?
EDC, or electronic data capture, is the system used to collect clinical data electronically, usually through eCRFs, patient-reported outcomes, surveys, and site-entered forms. A CDMS, or clinical data management system, is broader. It manages the clinical database, Database Specifications, CRF Annotation, edit checks, data validation, query resolution, Discrepancy Management, Source Document Verification, adverse event data, coding, reconciliation, database lock, and analysis-ready exports. Many modern platforms combine EDC and CDMS capabilities, but medical device teams should evaluate the full data management workflow and not just form capture.
What software is used for medical device clinical studies?
Medical device teams typically use validated clinical data management software or EDC software that supports device-specific study workflows. The right system should support ISO 14155, 21 CFR Part 11, audit trails, eCRFs, adverse events and device deficiencies, PMCF, eConsent when needed, patient- and clinician-reported outcomes, role-based monitoring, database lock, and secure exports for analysis and regulatory submission.
Let’s get started.
Table of Contents
![1]() 1. Why is clinical data management important?
1. Why is clinical data management important?
 1. Why is clinical data management important?
1. Why is clinical data management important?The simple answer is that good clinical data management is important because clinical trials involve human subjects and the devices studied may go on to be used by millions of other patients. The World Health Organization (WHO) estimates there are two million different kinds of medical devices on the market around the world, and it’s a near certainty that each and every one of us will interact with medical devices over the course of our lives.
Carefully collecting and managing data from clinical studies is part of keeping participants safe and ensuring that the study sponsor is able to draw accurate conclusions from the analysis of that data. As Article 62 of the European Union Medical Device Regulation (EU MDR) puts it:
Clinical investigations shall be designed and conducted in such a way that the rights, safety, dignity and well-being of the subjects participating in a clinical investigation are protected and prevail over all other interests and the clinical data generated are scientifically valid, reliable and robust.
From a business perspective, however, excellent clinical data management is imperative because that data is the backbone of submissions to regulatory bodies. Clinical data is the proof that a device is safe and effective for its end users, and should be allowed on the market.
How is clinical data used in the US and EU?
Typically, the devices that pose the highest potential risk must undergo clinical trials to collect the necessary data to prove their safety and effectiveness, though lower risk devices may also require clinical studies in certain cases.
In the US, approximately 10-15% of successful 510(k) submissions for Class II devices rely on data from clinical trials, and all Class III devices require extensive clinical trials to establish the reasonable certainty of their safety and effectiveness. FDA has even released a Q&A guidance document to help medical device companies understand the use of clinical data in submissions.
In the EU, Class IIb implantables and Class III devices will also require clinical investigations as part of their premarket regulatory pathway. Lower risk devices may also need clinical studies to determine their safety and effectiveness if there is not enough existing clinical evidence to prove it.
However, premarket clinical investigations are far from the only time you’ll need to collect clinical data. As you can see from the graphic below, clinical data will need to be collected throughout the lifecycle of your device.
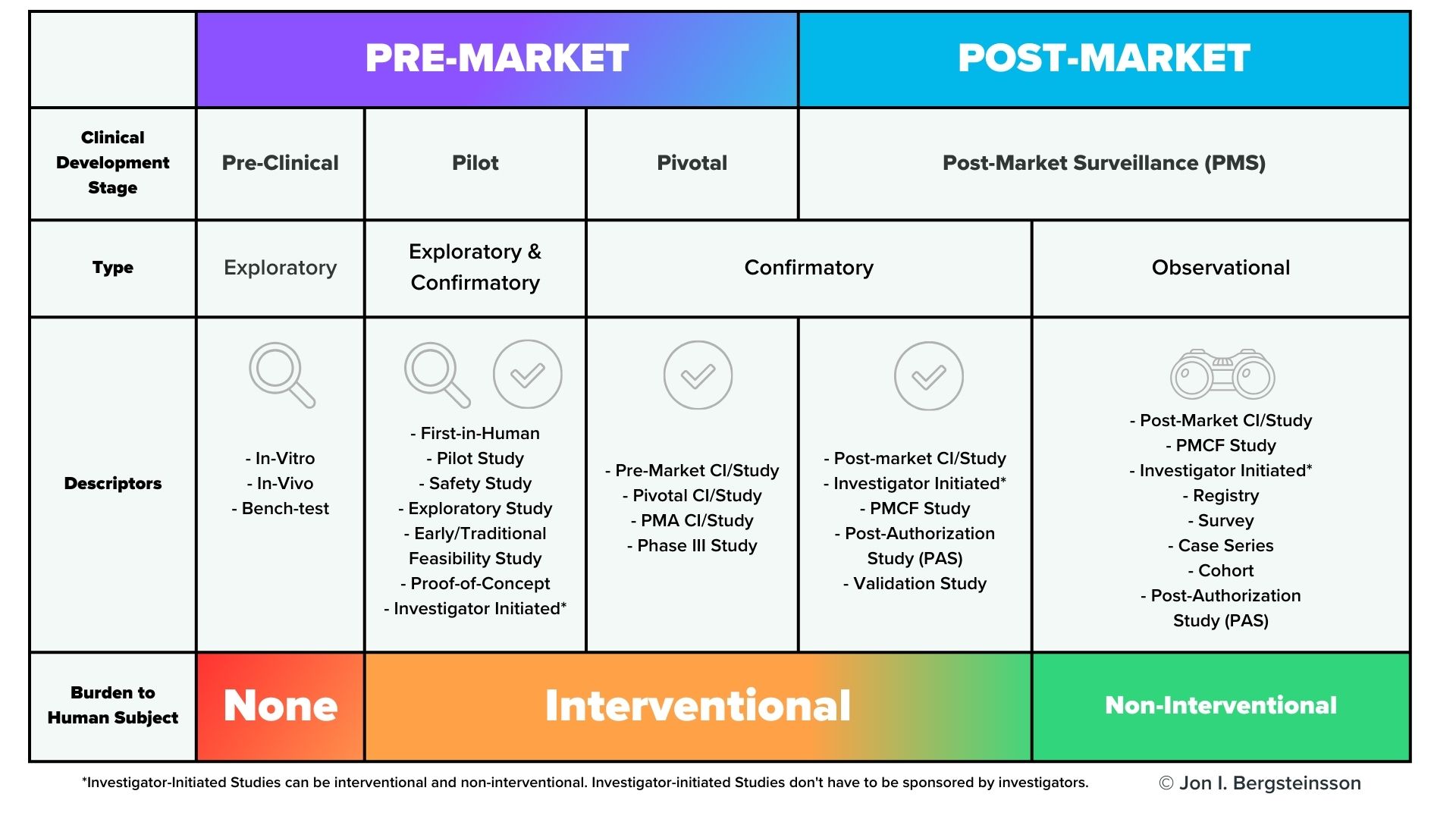 In the US, post-market surveillance of your device is a regulatory requirement, and FDA may also request a post-approval study of your device. In the European Union, EU MDR also has significant requirements for post-market surveillance that include the collection of clinical data, including potential Post-Market Clinical Follow-Up (PMCF) studies. You are also required to gather and evaluate all the clinical evidence for your device, which will then be published in a Clinical Evaluation Report (CER).
In the US, post-market surveillance of your device is a regulatory requirement, and FDA may also request a post-approval study of your device. In the European Union, EU MDR also has significant requirements for post-market surveillance that include the collection of clinical data, including potential Post-Market Clinical Follow-Up (PMCF) studies. You are also required to gather and evaluate all the clinical evidence for your device, which will then be published in a Clinical Evaluation Report (CER).
Keep in mind that the CER is a living document, and must be updated regularly. For Class III or Class IIb implantable devices, the CER must be updated at least once a year. For Class IIa devices, the CER must be updated at least once every two years.
Failure to do so will have consequences, as EU MDR also explicitly directs notified bodies to “pay particular attention to clinical data from post-market surveillance and PMCF activities undertaken since the previous certification or re-certification, including appropriate updates to manufacturers’ clinical evaluation reports.” (Annex VII, Section 4.11). Given the high priority that regulatory bodies place on clinical data, your ability to collect and manage data from clinical studies is paramount to getting your device on the market and keeping it there.
![2]() 2. How is clinical data collected?
2. How is clinical data collected?
 2. How is clinical data collected?
2. How is clinical data collected?For medical devices, clinical data collection happens throughout the entire lifecycle of the device, from early development all the way through the post-market stage. The graphic below provides another view of the various points in time during the device lifecycle that you’ll need to collect clinical data.
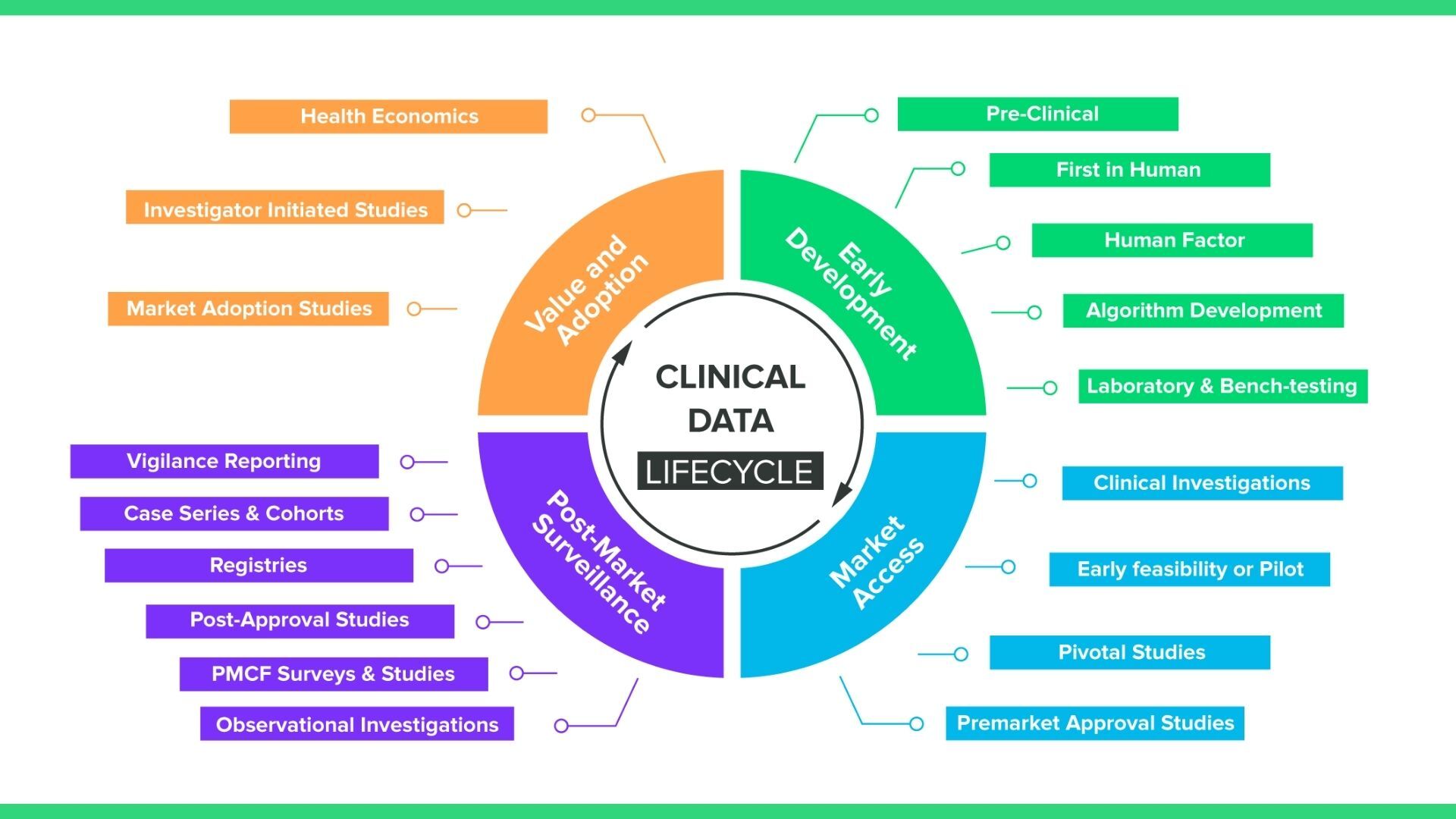
What you may notice is that while this graphic includes standard pre-market clinical investigations, it also includes a number of data collection methods, like registries and PMCF surveys, that are observational rather than interventional. What this means in practice is your EDC solution will require a number of different mechanisms for collecting clinical data, including:
- Case report forms (CRFs)
- Patient reported outcomes (PRO)
- Ad hoc data collection
- Post-market surveys
Case report forms (CRFs)
In clinical investigations, investigators traditionally use case report forms to collect data from participating patients, including patient characteristics and demographic data, adverse events, and the results of experimental treatments.
Paper-based CRFs have been standard practice in clinical investigation for decades, but medical device companies in recent years are increasingly choosing electronic CRFs, (eCRFs). This is due to much higher efficiency and a lower rate of errors when using eCRFs.
The reduced study time associated with eCRFs likely stems from the instantaneous access sponsors have to eCRF data. With paper CRFs, the physical forms have to be shipped somewhere and reentered in an electronic system. That builds a lag into any system that uses paper—sponsors have to wait until they get the data to do anything with it.
Speed is also related to another benefit of eCRFs: improved data quality. In a modern electronic data collection (EDC) system, fields will be pre-validated, meaning sponsors can set a data range for a given input. If an investigator enters a number that is outside of the data range, the system will alert them and may prevent them from moving on. So, if a physician who has been working for the past ten hours sits down to enter patient data and mistakenly adds an extra zero to one of her entries, the electronic form will catch the mistake.
And by following a few simple best practices for eCRFs, manufacturers can maximize the benefits of electronic case report forms and simplify data collection.
Patient reported outcomes (PROs)
Patient-reported outcomes are just what they sound like: data that the patient reports themselves (as opposed to the CRFs, which are filled out by site staff). PRO data has not always been the highest priority for manufacturers, but it is becoming increasingly important to medical device clinical trials. Regulatory strategy, reimbursement strategy, marketing, and more will all be affected to some degree by the data collected from patients.
As with CRFs, the best option for collecting PROs is now electronic. ePRO collection in clinical trials allows patients to self-report through an electronic device, such as a mobile app on a smartphone or tablet. This approach has several advantages over using paper forms:
- Instead of paper forms, patients can use the phone, tablet, or laptop they’re already comfortable with.
- Reminders can be sent using email or SMS to improve response rates.
- If the timing of patient data entry is a factor, ePRO software can make use of the notification features that devices already have to remind patients to fill out the form.
- Patients can fill out their ePRO questionnaire at home, on the go, or in the waiting room before seeing their physician.
- ePRO also makes use of data ranges and alerts that help keep patients from entering erroneous data that needs to be cleaned up later on.
Because of these advantages, studies have shown that ePRO response rates are significantly higher than paper forms and are also more accurate.
Post-market surveys
Post-market surveys are especially important for EU MDR’s post-market surveillance requirements, but they may also be required by FDA in the US. Post-market surveys are “observational”, meaning they use non-interventional methods to collect data. Here’s the difference:
- In interventional studies, such as a pivotal study, someone is actively recruiting participants. For example, a physician may ask a patient who may benefit from a certain device if they would like to volunteer for that study. In other words, they are intervening in the normal clinical pathway the patient would follow.
- In non-interventional studies, there is no intervention in the clinical pathway— merely observation. For example, a physician prescribes a treatment they believe the patient needs (the normal clinical pathway), and then asks the patient if they would agree to share the data related to their treatment as part of an observational study.
For these observational studies, which may be following subjects over long periods of time, it’s important to have a post-market survey method that is pre-validated and compliant with Good Clinical Practice (GCP).
Ad-hoc data collection
Finally, we have a method of data capture that is often forgotten: ad-hoc collection. As the name implies, ad hoc data collection doesn’t happen at set times—it happens when necessary or needed. This can make it difficult for manufacturers to collect this data in a GCP-compliant manner.
However, a tool like Greenlight Guru Clinical’s Cases module allows clinical staff to easily deliver data either during or after the application of a medical device in practice. This ensures that the data is fresh in a clinician’s mind and minimizes the burden of providing the data.
![3]() 3. What are best practices for clinical data collection and management?
3. What are best practices for clinical data collection and management?
 3. What are best practices for clinical data collection and management?
3. What are best practices for clinical data collection and management?Clinical data collection and management may feel like a complex and difficult undertaking.
However, with the right plan, the right tools, and a close eye on compliance, there’s no reason why you can’t get the high-quality clinical data you need from your next study. Here are the best practices we recommend for clinical data management.
Plan ahead for your clinical data management setup
It’s one thing to declare yourself ready to start a study and begin collecting data. It’s another thing entirely to do it. Most clinical studies are plagued by a lack of planning or foresight that ends up delaying the study timeline, and may even affect the quality of the clinical data obtained from the study.
However, when it’s done correctly and planned ahead of time, clinical data collection and management can be done without adding burdens to existing workflows. You can download our in-depth guide to planning a clinical data management setup, but here is a brief overview of the key steps you’ll need to take:
- Start with roles. Before you begin working on anything, assign roles and responsibilities so that everyone knows who is carrying out what part of your data collection and management process. Answer questions like:
-
- Who is responsible for setting up the data management solution, including forms, processes, and questions?
- Who should be responsible for translating?
- Who should be involved in testing your data management setup?
- Who will train study sites in data entry?
-
- Create a hypothesis. Every study requires a hypothesis and one or more study endpoints that will prove or disprove that hypothesis. Every endpoint can be defined by one or more variables, and these variables will need to be collected using forms or questionnaires.
- Sketch your plan. Here, you’ll consider how and when to collect the data for your variables. Depending on the study, this can happen all at once or at several times throughout the study pathway. Creating a visual timeline of your study and data collection points—literally sketching it out—is a great way to plan the study and will likely surface some issues or questions you haven’t already considered.
- Focus on “need-to-have” data. Many studies will initially be designed to collect every conceivable data point, because the sponsor feels that the more data they can collect, the better. In reality, the opposite is more often true. Collecting too much data can obscure the aim of your study, lengthen timelines, alter workflows, and even sour relationships with clinicians and study sites. Always ask yourself why you need a specific variable and how it will help you prove your hypothesis.
- Zero in on terminology. Standardizing the terminology and concepts you’re using before you design your forms and questionnaires will eliminate a potential source of confusion during data collection and analysis. This isn’t limited to medical terminology, either. All terms and concepts used in forms and questionnaires should be standardized and consistent to avoid any misunderstanding.
- Assign the data entry role. Data entry is traditionally mostly done by study personnel at the site. But patient reported outcomes (PRO) may be necessary to get the best data for your endpoints. Since data capture now happens to a large extent on digital devices, you may want to look into ePRO solutions that allow patients to answer questions in the comfort of their home and during their leisure, as these tools have been shown to raise subject compliance significantly.
- Take the time you need for testing. Everyone wants to get started on their study as quickly as possible, but to minimize errors in your actual study, you need to allow yourself enough time to test your data management setup—and test it more than once. Often, one test over the course of a single week is simply not enough. The first test will reveal errors or mistakes, and you’ll need at least one more test to ensure that you’ve fixed those mistakes properly, and that the fixes haven’t created more issues.
For smaller projects, i.e., studies or projects that include 1-15 forms or questionnaires, one or two sites, and under 30 participants, we recommend that set up should start no later than a month before data collection starts. If your study is bigger than that, we advise you to consult with your data management vendor as larger studies will be highly influenced by the solution you choose.
Ensure you’re complying with all relevant regulations and standards
Clinical data must be collected and managed in accordance with Good Clinical Practice (GCP) and a number of other regulations and laws within both the US and EU.
ISO 14155:2020 and Good Clinical Practice
Good Clinical Practice is a set of ethical and scientific quality standards for designing, conducting, recording, and reporting trials that involve human subjects. GCP is an internationally recognized standard, and ensures that the rights, safety, and well-being of subjects are protected and that all clinical data is credible.
ISO 14155—Clinical investigation of medical devices for human subjects— Good clinical practice, most recently updated in 2020, provides the standard requirements for medical device clinical trials to comply with GCP. The entire standard is required reading if you’re undertaking a clinical investigation of a medical device, but as far as data management goes, you’ll want to pay close attention to section 7.8—Document and data control.
ISO 14155:2020 requires any electronic system be validated “in order to evaluate the authenticity, accuracy, reliability, and consistent intended performance of the data system.”
So, if you’re using an electronic data capture (EDC) system, you will need to comply with procedures outlined in section 7.8.3, which include requirements to:
- Verify and validate that the requirements for the electronic clinical data system can be consistently met
- Ensure attributability, completeness, reliability, consistency, and logic of the data entered
- Ensure that data changes are documented and an audit trail is maintained
- Maintain a security system to prevent unauthorized use of data, both internally and externally.
And while that may seem daunting, with Greenlight Guru Clinical, you’ll have a system that comes pre-validated to all the requirements in ISO 14155:2020. That includes out-of-the-box compliance with the requirements in Section 7.8.3 and a suite of compliance document templates available to all customers.
The FDA’s 21 CFR Part 11 requirements
If you are using EDC software in the US, you’ll also need to comply with the requirements in 21 CFR Part 11, the FDA’s regulation on electronic documentation and electronic signatures. The regulation is broken up into three sections:
A. General Provisions
B. Electronic Records
C. Electronic Signatures
The data and records generated by clinical trials, as well as any electronic signatures (such as those used for eConsent) fall under the purview of 21 CFR Part 11. The goal of the procedures and controls outlined in Subpart B are to ensure that electronic records maintain their:
- Authenticity
- Integrity
- Confidentiality (when appropriate)
- Irrefutability (i.e. “the signer cannot readily repudiate the signed record as not genuine”)
The requirements for electronic signatures are found in both Subpart B and C. The electronic signatures requirements will be particularly relevant to you if you use eConsent in your clinical trials. Clinical trial managers may find gathering information digitally is safer and simpler than using physical documents—but that means the system they use to capture and store that data, will need to comply with the requirements in Part 11.
EU GDPR and HIPAA
In addition to regulations and standards surrounding Good Clinical Practice, you’ll also need to be aware of and comply with data protection regulations, like the European Union General Data Protection Regulation (GDPR) and The Health Insurance Portability and Accountability Act of 1996 (HIPAA) in the US. Due to the introduction of GDPR in 2018, there are a number of different obligations that MedTech companies must fulfill when conducting clinical trials in the EU, including:
- GDPR states that a clear and documented consent must be acquired from all data subjects in order to process their information.
- Medical device companies, or clinical trial sponsors, must now identify the data to be processed, where it will be transferred to, who is processing it, what it will be used for, and which risks are involved. All of which must now be included in a separate informed consent (not the protocol-specific consent).
- Organizations that process and manage clinical trial data must now conduct data impact assessments (DIA) on both electronic and hard copy data. A data impact assessment should cover what the data is used for, how it’s managed, and what action is needed to mitigate any risks.
- Sponsors are also required to appoint a Data Protection Officer (DPO) which shall take part in managing and documenting many of the activities that surround data and information processing. In addition, the DPO will also act as the main interface to the company if there are any data breaches or inbound inquiries. The DPO can either be an external hire or a current employee who you train for the role.
In the US, the Health Insurance Portability and Accountability Act of 1996 was passed to create national standards for the protection of sensitive patient health information from being disclosed without a patient’s consent or knowledge.
The three main HIPAA rules regarding Protected Health Information (PHI) in the US are:
- The Privacy Rule (Part 164 Subpart E): This rule safeguards the privacy of an individual’s health information and gives patients control over how their personal health information is used and disclosed, including the right to acquire a copy of their records.
- The Security Rule (Part 164 Subpart C): This rule establishes national standards for the security measures covered entities must take to protect electronic health information they create, receive, use, or maintain.
- The Breach Notification Rule (Part 164 Subpart D): This rule requires covered entities and their business associates to provide notification if there is a breach of unsecured protected health information.
In the US, sponsors of a medical device clinical trial will need to abide by all three of the HIPAA rules (Privacy, Security, Breach Notification), but the Privacy Rule has the most immediate impact on research. The Privacy Rule defines research as “a systematic investigation, including research development, testing, and evaluation, designed to develop or contribute to generalizable knowledge.”
When it comes to research, the Privacy Rule is meant to protect health information that could identify individuals while also making sure that researchers can access the data they need. You can find more information on complying with both data privacy regulations during clinical investigations in this article covering HIPAA and EU GDPR.
Use the right tools for your clinical data collection
Given the extensive planning and the multitude of regulations and standards you’ll need to abide by, managing your clinical data may feel daunting. The truth is that the difficulty or ease of collecting and managing clinical data often comes down to the data management solution you choose.
For example, clinical data collection and management is often hampered by the use of paper forms, Excel spreadsheets and other general-purpose software that leads to data entry errors, incomplete forms, and even non-compliance with regulations.
That in turn leads to data “cleaning” on the back end that can take an enormous amount of time and resources to perform, or worse yet, throwing out the data set entirely and starting over with renewed attention to compliance. And keep in mind, the regulations and standards I just went over are not suggestions. You must follow Good Clinical Practice in your study if you want that data accepted by any regulatory body. You must stay compliant with laws around data privacy, like GDPR, if you want to be able to access and use your data once a study is over.
To generate the type of high-quality clinical data that regulatory bodies will require to approve your submission or keep your device on the market, you will need to stay compliant with the requirements of all relevant regulations and standards.
The easiest way to stay compliant is by using electronic data capture software that offers built-in compliance with the requirements of ISO 14155:2020, FDA and EU regulations and customizable compliance document templates.
![4]() 4. Pitfalls to avoid in clinical data collection and management
4. Pitfalls to avoid in clinical data collection and management
 4. Pitfalls to avoid in clinical data collection and management
4. Pitfalls to avoid in clinical data collection and managementSometimes knowing what not to do is just as valuable as understanding best practices, and this is definitely true when it comes to clinical operations. Below I’ve listed some of the most common mistakes we see in clinical data management.
Starting off with paper
Collecting and maintaining clinical data on paper or Excel is perhaps the biggest mistake that MedTech companies make when they begin their clinical activities. That’s because starting with paper sets you up for failure in a number of ways.
For one thing, it’s challenging to ensure data integrity—meaning sufficiently documenting all aspects of data entry, data edits, and ensuring the data has not been modified, erased, or tampered with in any way—when you use paper records. And Excel simply is not built with the level of permissions or access management found in a pre-validated EDC solution.
Beyond data integrity, companies that use paper or spreadsheets also run into problems with project management. Clinical trials often happen at multiple sites, and without a firm plan for centralizing your data collection, it can create data fragmentation—especially if the methods for each entry location are different. This makes it extremely difficult to do things like:
- Set and monitor study milestones
- Identify and adjust for risk events
- Document the completion of tasks
- Automatically initiate tasks based on the completion of others
Finally, paper and Excel make it difficult (if not impossible) to comply with the regulations we talked about earlier. If we look to section 7.8.3 of ISO 14155:2020, we see that if an electronic system is used as the primary location of document storage and filing, the system must be validated in order to evaluate authenticity, accuracy, reliability, and consistent intended performance of the system. That means that whatever software used for storing clinical data needs to be validated, something that is nearly impossible to do with a general purpose system like Excel.
Clause 6 of ISO 14155:2020 covers clinical investigation conduct, and the good clinical practices outlined there are intended to ensure that the investigation maintains strict accountability and tight controls over documentation and recording. That means privacy must always be maintained and data should be protected against unauthorized access—two requirements that neither paper records nor Excel spreadsheets can guarantee.
Forgetting the patient’s perspective
Device manufacturers understandably tend to focus their attention on their device when they are performing clinical activities. They’re trying to understand how the device is performing, and it’s exciting to see it finally put to use helping patients.
However, it’s important not to lose sight of the central element of every clinical study: the patient.
Data from case report forms is still incredibly useful, but it’s important that you consider the patient’s point of view, as well, and collect it via patient reported outcomes. Aside from keeping the patient at the center of your study, PROs are a great source of data on your device and regulatory bodies and competent authorities place an emphasis on this data.
The need for PRO data is better known today, but there are still too many study sponsors who forget to include it. The data is typically not difficult to collect (if you’re using the right tool), and it’s an important part of the clinical data you need to collect to ensure the safety and effectiveness of your device.
Mixing clinical data collection methods
Additionally, mixing clinical data collection tools (using paper CRFs, but ePROs and recording it all in Excel) is another method I would urge you to avoid. The problems are the same as doing it all on paper or Excel, but with even more room for error.
One study found that when manually entering data into complex spreadsheets, the probability of human error was 100%. In a situation where the integrity of your data can be the difference between regulatory approval and denial, there is simply too much on the line to be manually entering data from different sources. Also, combining data collection tools often leads to compatibility issues.
Clinical data collection tools may have different file formats, data structures, or APIs that make them incompatible with each other. For example, one tool may collect data in a specific format that cannot be easily integrated with another tool.
Mixing informed consent and data consent
As we discussed briefly earlier in the guide, if you are pursuing clinical trials in the EU, you’ll need to obtain both informed consent (for participation in the study) and data consent (for sharing data with third parties, in accordance with GDPR).
These two types of consents often get mixed up or, just as bad, get combined into a single consent form. We see this happen a lot during investigator-initiated studies, where the investigators don’t have legal resources on hand to help them navigate the different types of consent. This can lead to extreme cases where a study went on for years, and at the end of it, the manufacturer of the device had no legal access to the data that was generated from the study.
Simply put: you need to obtain both informed consent and data consent, but you need to do so separately. You cannot use the same form for both.
Not using MedTech-specific clinical data management software
It’s also important to understand that not all EDC software or clinical data management software is built for MedTech companies. Many platforms were originally built for pharmaceutical trials, and there are significant differences between medical device studies and pharma studies.
From a functional standpoint, devices are often studied by observing how they are used in real clinical settings. As a result, device studies may be smaller, more iterative, more observational, and more dependent on input from healthcare providers, physicians, investigators, patients, and other users.
That creates a different data management problem. MedTech teams do not just need forms. They need a controlled Data Management System that can support the key activities of a medical device study, including eCRFs, ePRO/eCOA, PMCF surveys, adverse events, device deficiencies, Source Document Verification, audit trails, Discrepancy Management, query resolution, Database Specifications, CRF Annotation, Data Validation Plan execution, and database lock.
Generic CDMS and EDC platforms tend to break down for medical device teams when they treat data capture as the whole problem. For example, a Clinical Data Manager should be able to see missing data, open queries, unresolved discrepancies, coding issues, reconciliation status, monitoring status, and database lock readiness in one controlled workflow. A Clinical Research Associate should be able to document Source Document Verification and monitoring status without relying on spreadsheets, emails, or disconnected trackers.
Generic systems can also create problems during regulatory inspections. Inspectors and reviewers may need to understand who entered data, who changed it, when it changed, why it changed, which source was reviewed, which discrepancy was resolved, and which dataset was locked. If those records live in multiple systems, inspection readiness becomes manual and fragile.
Additionally, clinical data collection in device studies and clinical operations for devices is conducted around the whole life cycle of a device, from early-stage studies to market approval, post-market surveillance, and PMCF.
At the end of the day, the most effective solution is the one that is built with your specific problem in mind. An electronic data capture and clinical data management solution like Greenlight Guru Clinical, created specifically for MedTech companies and their unique needs, is what medical device companies should use for more efficient and effective data capture, data management, regulatory compliance, and clinical evidence generation
![5]() 5. Outsourcing clinical operations to a third-party
5. Outsourcing clinical operations to a third-party
 5. Outsourcing clinical operations to a third-party
5. Outsourcing clinical operations to a third-partyGiven the complexity and unique nature of medical device clinical trials, many MedTech companies will choose to work with a contract research organization (CRO). In fact, in Greenlight Guru’s 2024 State of the MedTech Industry Report, 70% of respondents told us they were going to outsource at least some of their clinical activities to a contract research organization (CRO) or consultant this year.
Some CROs are full-service, meaning they help with every stage of clinical operations, from site selection and participant recruitment trial monitoring and data management, while other CROs and consultants may focus on specific elements of the process like medical writing. Essentially, CROs can help your company with a wide range of services, throughout all stages of your clinical activities. It just depends on your needs.
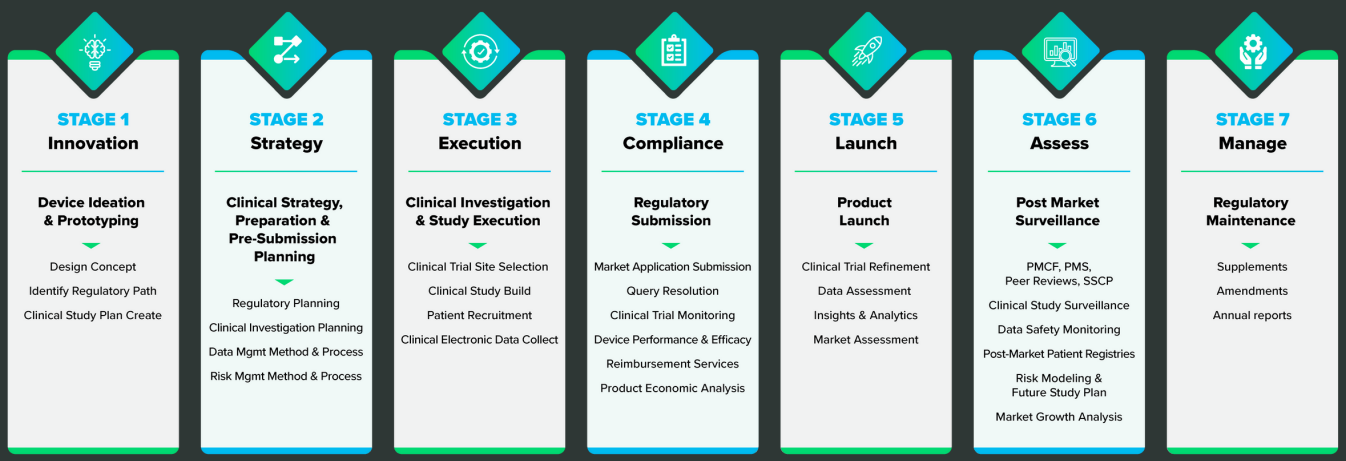
The choice of a CRO will revolve around your company’s capacity and experience with clinical operations, but there are some things to consider if you’re thinking about using a CRO or consultant. Here are a few steps you can take to help determine what you’ll need from a CRO:
-
Define your requirements (consider the SMART approach). In other words, your requirements should be Specific, Measurable, Attainable, Relevant, and Time-bound. This will help ensure that when you present your needs to a specific partner, they’ll quote you for what you need and nothing more or less.
-
Map which requirements can be handled in-house. Many internal clinical teams are fairly lean, and you’ll need to take a hard look at internal resources available to you. Consider whether you want to bring on more staff internally, or if you’d rather outsource more of the work.
-
Document results and refer back to your initial requirements. Once the process is underway, it’s important to track progress and continually assess whether your initial requirements are being met. Understanding your partner’s performance will help you course correct, if necessary, and help you decide whether this partner is the best fit for future clinical activities.
For a more in-depth method of determining your requirements and preferences for a CRO, download our free CRO Requirements Cheat Sheet, which covers everything from site access to cost structure and data access.
If you’re looking at using a CRO for any part of your upcoming clinical operations, I’d highly encourage you to check out the Greenlight Guru Partner Directory, where you can find many full-service and service-specific CROs to help you carry out your medical device clinical trials.

6. Choosing your clinical data management software
Even after finding a CRO, if you choose to use one, you’ll still be faced with a critical choice: what tools to use to collect, manage, review, clean, analyze, export, and retain the clinical data from your device’s study.
Remember, while your CRO may have preferences on an EDC solution—perhaps one they’ve used before—the choice is ultimately yours. This is your study and your data, and you are the one who decides how that data will be collected and managed. As I noted earlier, I would highly recommend you do not attempt to do your clinical data collection and management on paper or a general-purpose tool like Excel. The stakes are too high and the opportunity for errors in data entry, data cleaning, Discrepancy Management, and regulatory compliance are too great.
What you should really be looking for is a comprehensive clinical data management software solution that allows you to collect and manage your clinical data all in one place. The unique aspects of medical device clinical studies mean that you ideally want software purpose-built for MedTech companies, not a generic EDC or CDMS platform that forces your team to adapt device-specific workflows to a pharma-first system.
What clinical data management software does
Clinical data management software is the operational system behind a medical device study. It connects the protocol, case report form design, clinical database, data validation plan, monitoring activities, adverse event handling, reconciliation process, and database lock into one controlled workflow.
For medical device sponsors, the goal is not just to collect information. The goal is to produce complete, accurate, attributable, legible, contemporaneous, original, and consistent clinical data that can withstand internal review, regulatory inspections, and submission scrutiny.
Study design and Database Specifications:
The system should help the sponsor and Clinical Data Manager translate the protocol into Database Specifications, visit schedules, eCRFs, field logic, edit checks, required fields, controlled terminology, CRF Annotation, and reusable study templates.
Clinical data capture:
The system should collect data from sites, investigators, clinicians, patients, Data Entry Associates, and other users through eCRFs, ePRO/eCOA, PMCF surveys, registries, ad hoc data collection, and post-market workflows.
Discrepancy Management and query resolution:
The system should identify missing, inconsistent, out-of-range, duplicate, or illogical data. It should also allow the Clinical Data Manager to generate and track queries, assign owners, document responses, resolve discrepancies, and preserve a complete record of how data issues were handled.
Data Validation Plan execution:
The platform should support predefined validation rules, edit checks, field dependencies, range checks, visit-window checks, and review workflows that help the Clinical Data Manager detect problems before they delay analysis or database lock.
Source Document Verification and monitoring:
The Clinical Research Associate, monitor, sponsor, and site team should have a controlled way to review source data, document monitoring status, track open issues, and verify that clinical data entered in the system aligns with the source record.
Adverse event and safety data management:
The system should capture adverse events, serious adverse events, serious adverse device effects, device deficiencies, relatedness, severity, follow-up, reportability, and, when applicable, medical coding using the Medical Dictionary for Regulatory Activities or another approved terminology workflow with standardized codes.
Audit trails and regulatory compliance:
The system should maintain role-based permissions, electronic signatures, precise timestamps, record-change history, user accountability, and exportable audit trails. This is critical for ISO 14155, 21 CFR Part 11, GCP, regulatory inspections, and data integrity expectations.
Database lock and Database Locking:
The system should support a controlled database lock process after data entry, query resolution, coding, reconciliation, Source Document Verification, adverse event review, and final quality control are complete. Once the clinical database is locked, changes should be prevented unless the sponsor follows a controlled unlock, change documentation, and re-lock process.
Submission-ready exports:
The system should provide clean exports for statistical analysis, clinical evaluation, regulatory submission, PMCF reporting, and long-term study record retention.
Clinical data management software vs. EDC
Clinical data management software and EDC are closely related, but they are not always the same thing.
EDC software is the electronic data capture layer. It helps study teams collect clinical data through eCRFs, ePRO/eCOA, surveys, clinician-reported forms, and site-entered data.
Clinical data management software is the broader system used to manage the full clinical data lifecycle. It should support study build, Database Specifications, CRF Annotation, data validation, edit checks, query management, Discrepancy Management, Source Document Verification, medical coding, adverse event workflows, audit trail review, database lock, export, and archival.
How to evaluate clinical data management software vendors
Whether you are assessing Greenlight Guru Clinical or another clinical data management software platform, there are a few steps we recommend in evaluating and choosing a vendor.
There’s a good chance your organization will require you to formally evaluate and get quotes from multiple vendors. As you go through this process, a vendor evaluation checklist can be an excellent tool to keep track of different options and quickly compare them. Here are the key questions you should be asking every clinical data management software vendor:
- Is the system built specifically for medical device studies, or was it adapted from pharmaceutical trials?
- Which study types does the platform support: feasibility, usability, pre-market clinical investigations, registries, PMCF, post-market surveys, and ad hoc data collection?
- Does the system support the full clinical data management workflow, including eCRFs, ePRO/eCOA, adverse events, device deficiencies, Discrepancy Management, Source Document Verification, audit trail review, and database lock?
- How does the system support ISO 14155, 21 CFR Part 11, GCP, GDPR, HIPAA, and other relevant regulatory requirements?
- Is the software validated, and what validation documentation is included for sponsor review?
- How much work is required from the sponsor to perform software validation?
- Does the platform support Database Specifications, CRF Annotation, reusable study templates, field logic, edit checks, and version-controlled study builds?
- Can our team build and modify eCRFs without coding?
- How does the system support a Data Validation Plan?
- How does Discrepancy Management work? Can the Clinical Data Manager create, assign, track, resolve, and report discrepancies and queries?
- Can the Clinical Research Associate perform Source Document Verification and document monitoring status in the system?
- Does the system support adverse events, serious adverse events, serious adverse device effects, device deficiencies, severity, relatedness, follow-up, and reportability?
- Does the system support medical coding or standardised codes, including the Medical Dictionary for Regulatory Activities if required by the study?
- What does the audit trail capture? Does it show who changed data, what changed, when it changed, and why it changed?
- Does the system support precise timestamps, role-based permissions, electronic signatures, and inspection-ready records?
- How does database lock work? Does the platform support soft lock, hard lock, unlock approval, change documentation, and re-lock?
- What data exports are available for statistical analysis, regulatory submission, PMCF reporting, and long-term retention?
- How does the platform support data privacy and security?
- How quickly can the solution be implemented for a new study?
- Do you have medical device experts on your customer success team? If not, what is your experience with MedTech regulations and requirements?
- What support is available for the sponsor, Clinical Data Manager, Clinical Research Associate, Data Entry Associate, and study sites?
- Which statistical tools, reporting dashboards, and data export formats does your product support?
- What is the total cost of ownership, including license fees, implementation, validation support, custom programming, change requests, storage, and support?
- It’s also a good practice to speak with industry peers who are using the different solutions to get their points of view. When you compare vendors, evaluate the entire clinical data management workflow—not just the form-building or data capture experience
Device-specific requirements for clinical data management software
Medical device clinical studies have regulatory and evidence requirements that generic data capture tools are not built to handle well. Before selecting a vendor, evaluate whether the platform supports the requirements that matter most for device studies.
ISO 14155: ISO 14155 defines good clinical practice for clinical investigations of medical devices in human subjects. Your software should support controlled study conduct, credible clinical data, sponsor and investigator responsibilities, documentation, and data integrity expectations.
21 CFR Part 11: If electronic records or electronic signatures are used for FDA-regulated activities, the system should support Part 11 controls, including validation, record protection, access controls, secure computer-generated audit trails, electronic signature controls, and the ability to generate accurate, complete records for review.
Audit trails: Audit trails should show who created, modified, or deleted a record; when the action occurred; what changed; and why the change was made when required. Audit trails should be secure, time-stamped, retained with the study record, and available for review.
eCRFs and CRF Annotation: The system should help teams build protocol-aligned eCRFs, annotate fields, define variables, configure edit checks, and create a consistent clinical database structure before data collection begins.
Data Validation Plan: The platform should support the Data Validation Plan by enforcing required fields, logic checks, visit windows, range checks, conditional fields, and Discrepancy Management workflows that improve data quality before database lock.
Adverse events and device deficiencies: Medical device studies need structured safety data capture for adverse events, serious adverse events, serious adverse device effects, unanticipated adverse device effects, device deficiencies, severity, relatedness, follow-up, and reportability.
PMCF and post-market evidence: For EU MDR and lifecycle evidence needs, the system should support PMCF surveys, registries, patient-reported outcomes, clinician-reported outcomes, longitudinal follow-up, and reusable study designs for post-market data collection.
Database lock: Database Locking should be a controlled milestone. Before lock, teams should confirm that data entry, monitoring, coding, reconciliation, query resolution, adverse event review, and final QC are complete. After lock, the clinical database should be protected from uncontrolled changes.
Working with a clinical data management software vendor to establish a business case
Once you’ve found a vendor that you believe is the best fit for your company, work with them to build a business case for purchasing their solution that you can present internally to your executive team.
As you start to build the case together, it’s important to include both the benefits of the clinical data management software you’d like to purchase, as well as the ways it will affect the costs of running clinical investigations, managing data, resolving discrepancies, supporting regulatory compliance, preparing for database lock, and generating analysis-ready data. That might not be immediately recognized can include:
- Less travel time or no travel at all (with data monitoring and real-time data from studies).
- A reduction in time spent on data cleaning, data management, and prepping the data for analysis.
- Shorter study timelines due to faster study setup and reduced study closure time.
As you work with the sales team from the EDC vendor, ask if they have case studies that demonstrate cost-savings with their product. For instance, Oticon Medical—a long time Greenlight Guru Clinical customer and one of the top hearing aid manufacturers in the world—estimates they now complete their clinical studies 130-140% faster since switching to Greenlight Guru Clinical.
The Oticon team also estimated that their ROI on the purchase of Greenlight Guru Clinical was 136% after the first two years—and more than 270% after four years. Quantitative data like that, combined with the qualitative benefits of your chosen solution, will be critical to helping you build a strong business case that you can present with confidence to your executive team.
Establishing a transition plan
One final piece of an EDC business case that people often forget to consider is the transition plan. Also referred to as a project plan, the transition plan is the summary of how you intend to get from A to B—meaning from paper or your old EDC system to the new one. It should answer questions like:
- What are the steps involved in transitioning to the new EDC system?
- What kind of support will you get from the EDC vendor on the implementation?
- Who will need to be trained on the new software and how long will that take?
- Are there any risks involved in transitioning and what are you doing to mitigate them?
- What is the estimated timeline for the transition, and how does it interact with key dates like the start of a study or regulatory submissions?
- What are the specific data collection needs for any upcoming studies?
As with every robust system, there will be an onboarding process. Learn from the sales team how long that will take, and when it is best to begin if you need to start your study in the next couple of months.
With Greenlight Guru Clinical, medical device companies can set up their study within a couple of weeks and even faster depending on size or complexity. In some cases, our Customer Success team can even help with building parts of your first study to speed this process up.
If you’re just beginning the journey of purchasing a new electronic data capture or clinical data management software solution, I encourage you to check out our EDC buyer’s guide. It will walk you through the steps of evaluating software and getting executive buy-in for your purchase—whether you decide to use Greenlight Guru Clinical or not.
As you evaluate vendors, make sure your shortlist is based on the full clinical data lifecycle: protocol-aligned study build, Database Specifications, CRF Annotation, data capture, Data Validation Plan execution, Discrepancy Management, Source Document Verification, adverse event review, regulatory compliance, database lock, export, and inspection readiness.
Why Greenlight Guru Clinical is purpose-built for MedTech
Greenlight Guru Clinical is built for medical device clinical evidence, not retrofitted from a pharma-first trial model. It gives MedTech teams one platform for collecting, managing, reviewing, and using clinical data across the device lifecycle.
With Greenlight Guru Clinical, teams can support early feasibility studies, pre-market clinical investigations, PMCF, registries, post-market surveys, eCRFs, ePRO/eCOA, eConsent, adverse event reporting, and secure data workflows in one validated platform.
For lean MedTech teams, that matters. The Clinical Data Manager, Clinical Research Associate, sponsor, and study sites can work in a system designed around device evidence needs instead of forcing device workflows into a generic CDMS or EDC system. That helps reduce manual reconciliation, improve visibility, maintain audit-ready records, and move more efficiently from study build to database lock.
Greenlight Guru Clinical is especially well suited for teams that need to
- Build and reuse eCRFs without heavy custom programming
- Collect data from patients, clinicians, investigators, and post-market users
- Manage adverse events and device-specific safety workflows
- Support PMCF and lifecycle evidence collection
- Maintain audit trails, Part 11-aligned e-signatures, and ISO 14155-aligned workflows
- Track study progress and data quality in real time
- Prepare clean, controlled datasets for analysis and regulatory submission
If you are evaluating clinical data management software for a medical device study, prioritize the system that supports the entire data lifecycle: protocol-aligned study build, CRF Annotation, Database Specifications, data validation, Discrepancy Management, monitoring, adverse event review, database lock, export, and inspection readiness.
7. Final Thoughts![create business case]()

I’m not exaggerating when I say that a well-planned and perfectly executed study means nothing if you can’t collect, manage, review, clean, lock, and export the clinical data the study generates. Simply completing a study does not necessarily prove that your device is safe and effective for its end users.
Those assertions can only be proven through the data plus the integrity of the data you collect from your study. And nothing has a greater effect on your ability to gather and analyze high-quality clinical data than the clinical data management software you choose to help you accomplish that task.
The right platform should support the full workflow: eCRFs, ePRO/eCOA, adverse events, PMCF, audit trails, Discrepancy Management, Source Document Verification, Data Validation Plan execution, regulatory compliance, database lock, and analysis-ready exports.
When you choose to partner with Greenlight Guru Clinical, you’re not only getting a MedTech-focused toolbox for clinical data collection and management, you’re getting dedicated customer service from MedTech professionals who understand the unique nature of medical device clinical trials.
So if you’re looking for a partner who will be with you every step of the way, then Get Your Free Demo of Greenlight Guru Clinical.→
FREE eBOOK:
Ultimate Guide to Clinical Data Management in MedTech
Ultimate Guide to Clinical Data Management in MedTech






