.png?width=800&height=400&name=How%20To%20Use%20FDA%E2%80%99s%20Guidance%20on%20Post-Approval%20Studies%20During%20Premarket%20Approval%20(new).png)
In the US, post-market clinical studies are not required of every medical device manufacturer. However, FDA does have the authority to order post-market surveillance of any device, and for devices in the Premarket Approval (PMA) pathway, this may come in the form of a Post-Approval Study (PAS).
According to FDA’s guidance document, Procedures for Handling Post-Approval Studies Imposed by Premarket Approval Application Order, a PAS may be ordered “to provide reasonable assurance, or the continued assurance, of safety and effectiveness of an approved device”.
If your device requires a PMA application, there’s a decent chance it will also require a Post-Approval Study (PAS). If that’s the case, the FDA’s guidance document is an invaluable resource for understanding what will be required during a PAS, the important timelines for your study’s progression, and how you’ll communicate with FDA.
I won’t go through the guidance line-by-line, but in this article I’ve highlighted some of the most salient information for device manufacturers who may be ordered to do a PAS as part of their PMA.
What is a Post-Approval Study?
A Post-Approval Study (PAS) is one of the methods FDA may require for post-market surveillance of a device, and it’s typically reserved devices that are using the Premarket Approval (PMA) or Humanitarian Device Exemption (HDE) pathways. And the guidance document we’re discussing today is for those taking the PMA route.
A Post-Approval Study is exactly what it sounds like: a study that is carried out after FDA has approved your device to be placed on the US market. It’s used to provide more evidence for the safety and effectiveness of your device after it’s been approved.
The guidance states that FDA may order a PAS when the agency “has uncertainty regarding certain benefits or risks of the device, but the degree of uncertainty is acceptable in the context of the overall benefit-risk profile of the device at the time of premarket approval.”
In other words, they believe your device’s benefit-risk profile means it should be approved, but they have lingering question(s) about the device’s safety and efficacy that need to be cleared up via a clinical study.
What is your Post-Approval Study protocol and when is it reviewed?
The Post-Approval Study protocol is one of the first items you’ll need to agree upon before moving forward with a clinical trial of your medical device. The agency encourages manufacturers to “engage with FDA as early as possible in the PMA review to discuss the need for and details of a PAS protocol.”
As the guidance states, this is meant to be a collaborative process:
When a PAS is likely to be required as a condition of approval, sponsors and FDA should work collaboratively to establish a PAS protocol, enrollment milestones, and study completion timelines prior to PMA approval to help ensure that the PAS achieves its objectives and is completed in a timely manner.
You can find the full list of recommended elements in the PAS protocol in Section IV, Part A of the guidance document, but your protocol must include:
-
The device’s background (description, regulatory history, indications for use, etc.)
-
The purpose of the study
-
The objectives of the study
-
The study design
-
The study population
-
Your enrollment and recruitment plan
-
A statistically justified sample size calculation
-
Length of follow-up, follow-up schedule, plans to minimize losses to follow-up, and estimated follow-up rates
You should consider that last bullet point carefully. You aren’t going to get a do-over on your Post-Approval Study, and if you’re unable to follow up with subjects or can’t get accurate data in the form of patient reported outcomes (PRO), then you may not be able to satisfactorily answer the FDA’s questions about your device.
With Greenlight Guru Clinical, patients can report outcomes on their own devices, whether on-site or at home. The software can prompt them to answer via SMS or email at specific times, which often increases response rates enormously.
What’s the timeline for PAS Protocol Review?
In the FDA’s guidance document, the agency notes that there are a couple different timelines for the Post-Approval Study protocol review. The first method is simply to review the protocol and establish milestones for enrollment and timelines for completion at the time of PMA approval:
When a PAS is likely to be required as a condition of approval of the PMA, FDA intends to review the PAS protocol interactively with the sponsor during the review of the PMA and concurrent with the review of the premarket data.
However, if the Post-Approval Study protocol hasn’t been developed by the time of the PMA approval, FDA may approve the PMA with a Post-Approval Study outline (which includes objective and general study design, study population, study endpoints, sample size, length of follow-up, frequency of assessments, and enrollment milestones).
But you must then submit a PAS protocol for approval within 30 calendar days of the PMA approval. The agency intends to review the submitted PAS protocol within 30 calendar days after receiving it—meaning the PAS protocol review completed within 60 calendar days of the PMA approval.

What is the timeline for enrolling subjects for your Post-Approval Study?
One of the key timelines to know is for enrolling subjects in your study. FDA expects subject enrollment to progress in this manner:
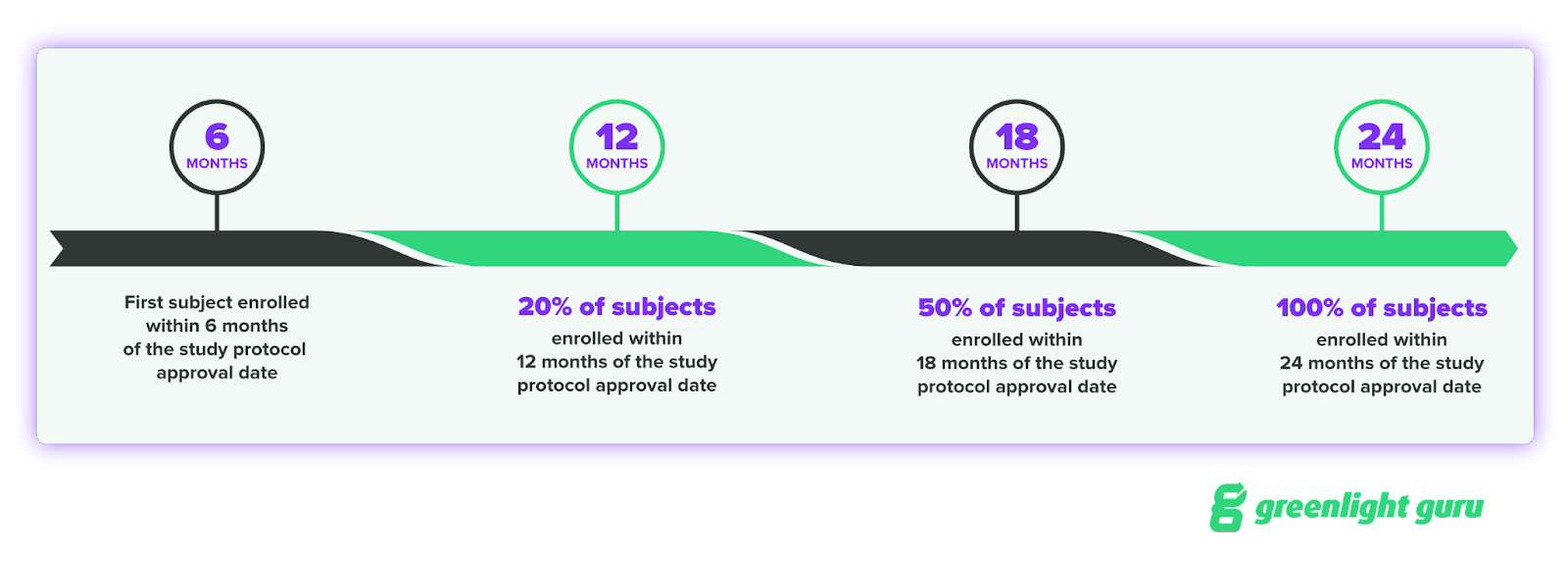
Six months is a relatively short timeline, and if you’re using paper or a clunky electronic data capture (EDC) system to set up your study, that six month period may not be enough time.
Greenlight Guru Clinical can speed up your time-to-enrollment with an intuitive, three-step study builder with simple, drag-and-drop elements, form validation, and customizable protocol designs. And because it’s pre-validated per ISO 14155:2020, you’ll spend less time worrying about compliance with medical device regulations and standards, and more time enrolling patients and obtaining the clinical data you need.
What are your Post-Approval Study Reports and what is the timeline for submitting them?
There are a few different types of Post-Approval Study Reports that you’ll be required to submit to FDA over the duration of your PAS:
FDA tracks and evaluates the conduct of a PAS through review of study reports submitted to the Agency. An interim report is a written report to FDA on the status of the PAS prior to its completion. Generally, FDA recommends submitting two types of interim reports: “Enrollment Status Report” and “PAS Progress Report.”
You can find a detailed list of the information that should be included in each report in Section VI of the guidance, but here is an overview of the different reports and their timelines:
-
The Enrollment Status Report is a document that shows your progress toward meeting the enrollment milestones that we discussed in the previous section.
-
The PAS Progress Report describes the status of the Post-Approval Study before completion and includes subject accountability, device performance, and safety and effectiveness data. If delays have occurred or are anticipated, you should notify FDA in your next PAS Progress Report.
-
There is also a Final PAS Report, which is the documentation of a completed or terminated Post-Approval Study.
What are the timelines for submitting your PAS reports?
The timelines for submitting these reports will not be exactly the same for every company, and the agency states in its guidance that “FDA intends for the PMA approval order to include a submission timeline for PAS reports.”
However, the timeline for the Enrollment Status Reports will likely be based on the deadlines for the subject enrollment milestones.
As for the PAS Progress Reports, FDA will generally require you to submit one every six months until all subjects have been enrolled. After that, they may be submitted annually until the Post-Approval Study is complete or terminated. It’s also possible that the timing of an Enrollment Status Report will coincide with one of the enrollment-period PAS Progress Reports, in which case you can combine the two to meet your reporting requirements.
The Final PAS Report is generally submitted “no later than three months after the study is complete (i.e., last subject’s last follow-up date), unless a different reporting schedule post study completion is agreed upon.”
Just remember that the timeline for these submissions is what FDA “generally” requires, and yours may be different. Your specific reporting timeline will be agreed upon with FDA prior to the start of your Post-Approval Study.
TIP: You can use the Post-Approval Studies Database to get a better understanding of previous and ongoing Post-Approval studies, such as PAS protocol parameters and reporting timelines.
Streamline your PAS with a MedTech-specific clinical data collection solution
If the thought of a Post-Approval Study sounds daunting, you’re not alone. Conducting a successful Post-Approval Study to collect the most relevant clinical data can be a challenge. The majority of clinical data collection tools out there have been designed to support pharmaceutical clinical trials—which are vastly different from post-market device studies.
Collecting data on a medical device in the post-market stage is more challenging than in pre-market studies, because people tend to forget that the device is still under observation. To optimize resources and streamline the data capture, you need to include an electronic data capture solution (EDC) that is designed specifically for this purpose.
Greenlight Guru Clinical was specifically designed to facilitate MedTech clinical operations and ensure compliance from the start with built-in ISO 14155:2020, EU MDR, and FDA requirements.
The platform contains a variety of data collection methods and tools to help you collect, manage and oversee the clinical data gathered in your PAS. This includes features like eCRF, ePRO, Surveys and more—all of which can be applied as traditional clinical studies, registries, case series and more.
Get Greenlight Guru Clinical today!
Clinical Electronic Data Capture (EDC) Software





