
What does it mean to create a risk-based CAPA process? How should companies go about it?
Create a CAPA risk assessment in 13 steps
Companies can create a risk-based CAPA process and be compliant with the latest revision of ISO 13485:2016 updates to ISO 13485. A key question many ask is, how do you convert what you have into being something “risk-based” and compliant with the new regulations?
Here are some steps to creating that process:
Free Bonus Guide: When to Use Different Tools for CAPA Analysis. Click here to download free PDF.
1. Know the inputs
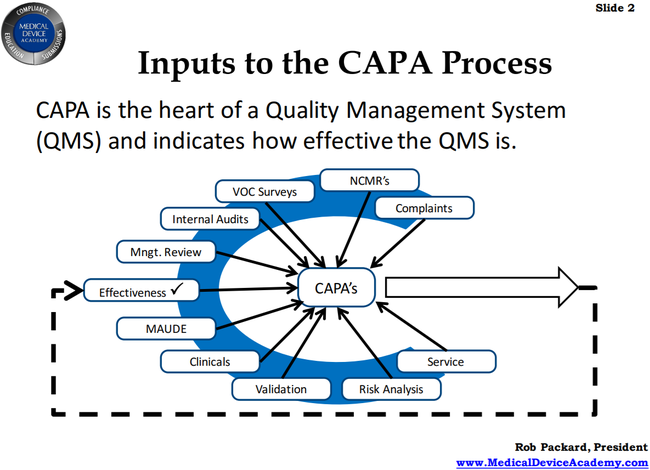
There are a lot of inputs to CAPA and it is meant to be the heart of your quality management system (QMS). as it helps you fix any problems. One of the things that the FDA expects is that you should have all of these various items inputting to your CAPA - all from one source is not good.
For example, if you look at the diagram below, MAUDE is an input, but that doesn’t mean just monitoring within your own company. It is expected that you would look at competitors and any issues they may have had in order to inform your own CAPA. This means that you might be implementing preventive actions, hopefully before you reached a situation where you needed to take corrective action.
2. Design a great form
Having a well-designed form helps you iron out the bugs and create something that is logical and step-by-step. When people don’t fill out forms correctly, it’s often because the form is not well-designed and has things out of order. Forms should include plenty of room and include all of the elements necessary for CAPA.
(Note: Looking to implement a risk-based CAPA process with a proven CAPA form design out-of-the-box, this is part of what companies can get with Greenlight Guru's postmarket workflows. Learn more and see it live here.)
3. Learn how to analyze CAPAs
One of Rob’s biggest pet peeves is when people say they “have to” include a 5 Whys analysis on their report.
He emphasizes that 5 Whys is just one method in a list of several possibilities to help get to your root cause of any issue. You might use it, but you might use others, even multiple methods to analyze CAPAs.
He recommends that you don’t have 5 Whys embedded into your form, rather that you have a range of tools you might use and that you know when to use them.
4. Understand “risk-based”
A common question asked by companies is why are we shifting to this risk-based focus? As Rob points out, it has always been there, however the 2016 update mentions the word “risk” many more times.
The point is that risk should be an underlying concern for all parts of your QMS. Companies should be conversant with the guidelines for quality system implementation and use the right tools to help them set up (such as Greenlight Guru for an all-in-one cloud-based system).
Rob recommends that, for a thorough guidance document, companies use “13485 Plus”, which is a Canadian guidance for quality system implementation. It mentions “risk” 60 times and is a useful tool. 14971 Plus is another he recommends - it contains the standard, as well as highlighting changes and bonus tools.
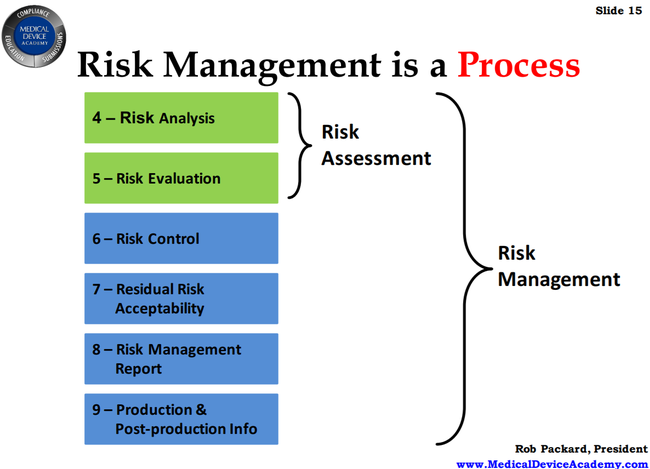
5. Know the risk management process
Rob shared a diagram he created on the risk management process, which you can see below. He highlights that in order to have a great risk management process, you need to pay attention to all parts and most especially that last piece - production and post-production information.
You need to be using this information to assess your process and understand whether you are managing risk effectively.
6. Use risk as a filter and prioritization tool
How does your company determine when a CAPA should be opened? The point is that not every incident should be a CAPA. Look for systemic issues or issues of significant severity as a guide.
Rob tells a story which highlights this. He was sitting next to someone whose company opened a CAPA every time someone was slow to respond to a pager alert. This could potentially mean a huge number of open CAPAs, an onerous task to go through. A different way to look at it would be one CAPA for all incidents of slow response, so that the company can get to the bottom of why the response might be slow.
Of course, any incident that could lead to significant harm should trigger an instant CAPA but you will have issues that occur that don’t fall into this category. Are they worth of a CAPA or not? Sometimes they will be managed through other processes (such as risk management) without needing to open a CAPA.
Myth Busting: Rob makes an important point here - there is no such thing as the “rule of three” that many companies seem to think exists. This is the idea that if something happens three times, it should be a CAPA. There is nothing in the FDA guidelines to indicate this rule, it’s just something that has caught on erroneously.
7. Use the right definitions
A couple of important points here:
- The word “mitigation” implies the elimination of risk and so has been removed from the standard. Risk can never really be eliminated, so we use terms like “control”, meaning we can reduce and monitor risks.
- The term “risk” as defined in ISO 9001 is completely different to how we use it in the medical device industry. Be sure not to use the 9001 definition, instead use 14971 as your definition.
8. Planning and documentation is required
The guidelines under 13485:2016 don’t have any significant changes for corrective or preventive actions, but one thing they do require is appropriate planning and documentation. The FDA will want to see your planning as far as attending to CAPA goes, so be sure to include this as part of your process.
9. Containment and correction
In parallel with any action you are taking to correct or prevent a further issue, you should be running containment to stop it from occurring while you’re investigating the issue.
In terms of corrections, an auditor might find a few contributing factors to the issue at hand - those are all going to be corrections, as are any that the FDA gives you in a 483 observation. Note that they will want you to go back over 2 years of data, which is potentially a lot of corrections to be made.
10. Have risk controls in place
There a few types of risk control, but here are a couple commonly used:
- Inspection - This is more of a containment method, but you can use it to gather metrics for data analysis. Factors like frequency and depth of inspection should all be risk-based decisions.
- Process validation - This is more proactive. How capable is our process in making things to inspect? Use data from your process validation to determine a risk-based approach to measurement.
11. Know your P1 and P2
The terms P1 and P2 are often confused by companies. They are found in the ISO definition of risk and defined as:
- P1 = the probability of a hazardous situation occurring.
- P2 = the probability of a hazardous situation leading to harm.
- P1 x P2 = overall risk (High, medium, low, based on parameters you will have set).
To be clear, you can get your P1 value from your process validation, however P2 data comes from the product being released into the field, or post-market surveillance. Sometimes you can get that information from clinical study, but post-market surveillance tends to be more reliable.
The point of this is that you need to determine how you will calculate risk scores for your CAPA process, particularly looking at the probability of the occurrence of harm.
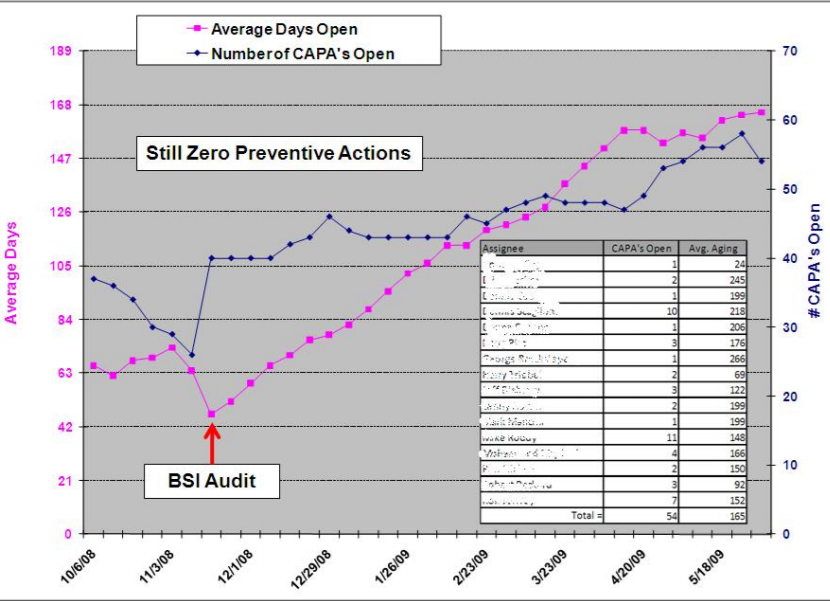
12. Monitor data
It’s important that you devise a process for monitoring your CAPAs (or use a software solution like Greenlight Guru that does it for you) and noting whether or not it is effective. Rob has used the method of monitoring the number of days a CAPA remains open vs. the number of CAPAs open to build a good overall picture, as seen below.
13. Plan a risk-based eQMS
When you plan your QMS, include monitoring and measuring in that plan and come up with a strategy for making your QMS risk-based. The guidelines state that management must ensure that the QMS meets ISO 13485:2016, clause 4.1 as well as quality objectives.
The integrity of the QMS must be maintained when any planned changes are made.
This raises another good point: If you are making any changes, then you should implement a training plan and check for competency.
Free Bonus Guide: When to Use Different Tools for CAPA Analysis. Click here to download free PDF.
Final thoughts
As you can see from this, much of your success or challenges with CAPA will come from how well you prepare ahead. A well-designed process will help you greatly, along with a good form to logically follow the process.
It’s a bit of homework for companies, learning the standards and terms, but putting that work in upfront can pay off by helping you to avoid drawn-out processes or audit findings later on.
(Note: If you're looking to implement a risk-based CAPA process at your device company that streamlines and connects with all your other quality processes out-of-the-box, I would recommend you check out our postmarket workflows. Learn more and see it live here.)




