








Marketing a device in the European market means complying with the numerous safety and technical requirements of the Medical Device Regulation (MDR). In practice, this can be an extremely challenging process, particularly for startups or companies who are new to marketing devices in Europe.
That’s why we’ve created this ultimate guide to break down the device class requirements under the European MDR. Along with instructions on how to classify and categorize your devices, you’ll learn exactly what you need to know for preparing technical documentation, conducting clinical evaluations, running post-market studies, and ensuring your device is safe and effective.
Without any further preamble, let’s take a look at classifying your device and what it means for your path toward CE marking and legal sale under EU MDR.
Table of Contents
 Classifying and Categorizing Devices Under MDR
Classifying and Categorizing Devices Under MDR
There are a number of different routes of assessment to obtain CE marking for your product, and the route you take depends on the risk class of your device under the MDR. In addition to the classification requirements detailed in MDR, the Medical Device Coordination Group (MDCG) published the guidance document MDCG 2021-24 as a simplified resource to help manufacturers determine the class of their medical devices under Regulation (EU) 2017/745.
The MDR designates four medical device classifications:
-
Class I
-
Class IIa
-
Class IIb
-
Class III
Each of these risk classes requires a different conformity assessment route, which will determine the steps you’re required to take for CE marking. The EU uses a rule-based system for determining the risk class of a medical device. In Annex VIII of the MDR, you’ll find 22 rules for classifying any medical device.1
The rules are divided into four sections, and the rules of each section apply to a specific category of devices.
-
Rules 1-4 cover non-invasive devices.
-
Rules 5-8 cover invasive devices.
-
Rules 9-13 cover active devices.
-
Rules 14-22 are special rules that cover devices that do not fall into the first three categories, such as nanomaterials.

Additionally, the duration of the device’s use is used to determine which rule(s) apply within a given category.
The three types of duration specified in the MDR are:
-
Transient: Intended for continuous use for less than 60 minutes
-
Short Term: Intended for continuous use for between 60 minutes and 30 days
-
Long Term: Intended for continuous use for more than 30 days
.png) Choose the rule(s) most applicable to your device in order to determine the risk class of your medical device in the EU. If several of the rules apply to your device, the MDR stipulates that you should use the strictest rule that results in the highest level of classification.
Choose the rule(s) most applicable to your device in order to determine the risk class of your medical device in the EU. If several of the rules apply to your device, the MDR stipulates that you should use the strictest rule that results in the highest level of classification.
I’ve determined my device classification — now what?
Overall, MDR is less focused on the pre-approval stage of medical device manufacturing, and instead, promotes a lifecycle approach to medical device regulation. This can be found in changing safety requirements and technical documentation.
The general requirement under the new MDR is that all currently approved devices must be re-certified to ensure their compliance with the new medical device requirements. Even medical devices that obtained CE markings under the previous directive must demonstrate compliance with the new EU MDR.
Additionally, manufacturers of Class IIa and IIb medical devices will need to review their existing clinical evaluations to determine whether they satisfy the updated regulations. Certain medical devices that were previously exempt from requiring clinical evaluations may now.
Skip to your medical device class:
![eu-device-requirements-class-I]() Device Class I Requirements
Device Class I Requirements
 Device Class I Requirements
Device Class I Requirements Class I medical devices in the EU have the lowest perceived risk. In many cases, the manufacturer can self-certify Class I devices without the involvement of a notified body. This risk class includes products like stethoscopes, bandages, or glasses.
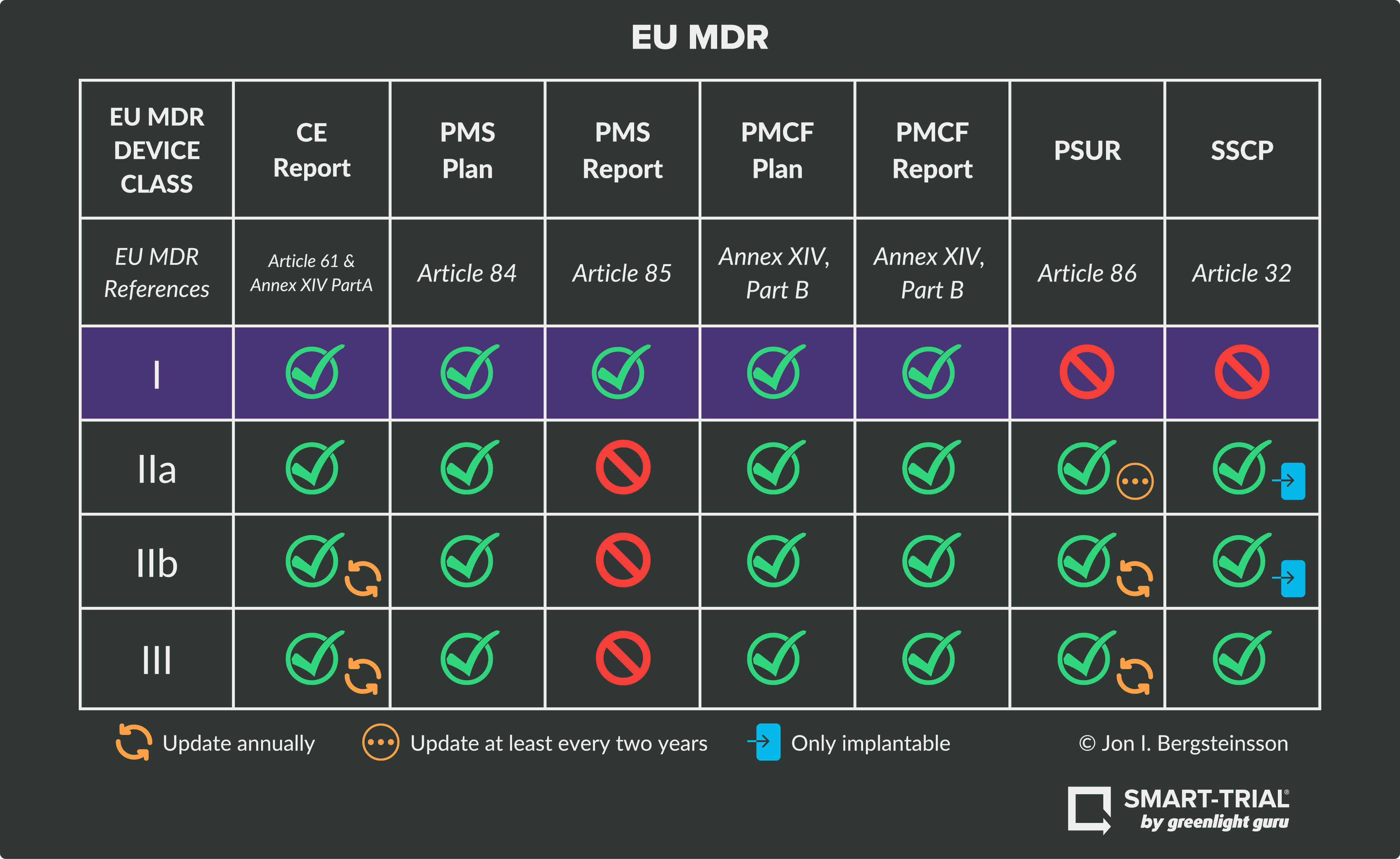
As seen in the graphic above, Class I devices must fulfill obligations and technical requirements for:
-
Clinical evaluation report (CER)
-
Post-market surveillance plan (PMS Plan)
-
Post-market surveillance report (PMS Report)
-
Post-market clinical follow-up plan (PMCF Plan)
-
Post-market clinical follow-up report (PMCF Report)
NOTE: Class I devices do not require Periodic Safety Update Reports or Summary of Safety and Clinical Performance submissions.
Without further ado, let’s jump into the specifics of each safety and technical requirement for your Class I device under EU MDR.
CE Report
What is it? The clinical evaluation report (CER) is documentation of the clinical evaluation that is required of every medical device sold in the EU. Its purpose is to prove your device performs as intended without compromising the safety of its end users.
How is it regulated under EU MDR? Article 61 of EU MDR requires every medical device manufacturer to document the clinical evaluation of their device in a CER.3 This requirement is expanded upon in Annex XIV Part A, which states:
The results of the clinical evaluation and the clinical evidence on which it is based shall be documented in a clinical evaluation report which shall support the assessment of the conformity of the device.
MDCG-2020-5 and MDCG 2020-13 are guidance documents for clinical evaluations, and contain instructions and specific requirements for the clinical evaluation report.5
How to comply? MDCG-2020-5 states that your CER should contain sufficient information for anyone reading it to understand the search criteria, the included data, and all assumptions made as well as all conclusions reached.
MDCG-2020-13 provides a sample table of contents, which indicate the CER should include:
-
Administrative particulars
-
Medical device name model and type
-
Manufacturer(s) name and SRN
-
Notified body
-
Type of assessment
-
Intended purpose
-
Check of clinical evaluation report authors
-
Reviewers involved in notified body assessment of the clinical evaluation
-
Device information, including:
-
Description
-
Classification
-
Clinical evaluation plan
-
Information materials supplied by the manufacturer
-
Common specifications and harmonised standards applied
-
Demonstration of equivalence
-
State of the art
-
Clinical literature review
-
Clinical investigations and related documentation
-
PMS, PMCF and the plan for updates
-
IFU, SSCP, labeling and other information supplied with the device
-
Summary of all available data and conclusions
-
Overall Conclusions
Lastly, the guidance states that the report’s contents shall be cross-referenced to supporting documents, and to their locations in your technical documentation.
When do I submit or update the CE Report? For Class I devices, the CE Report must be updated every 2-5 years. Because Class I devices are low-risk, the guidance allows for you to define your own update schedule based on the device’s intended use, frequency of usage, and overall level of risk. However, if at any point you receive new and pertinent information about your device through postmarket surveillance or new clinical evaluations, then the CER must be updated to reflect that.
How can Greenlight Guru help you manage the entire clinical evaluation process? A successful clinical evaluation report hinges on the quality of clinical data and the processes for collecting and analyzing it. To do so, researchers use Case Report Forms (CRFs) completed by investigators for each patient during the investigation.8
In the past, CRFs from a clinical study would be completed on paper, packed into boxes, and shipped from the study location to a secondary location for analysis. But today, a growing number of medical device companies are going paperless and using web-based Electronic Data Capture (EDC) systems to collect, store, and manage clinical data.9
EDC systems give clinical study managers the ability to streamline the data collection process, capture more accurate data, and enhance data security and accessibility - all while saving time and reducing the cost of completing a clinical study. EDC systems are also an essential pairing with a well-developed quality management system to ensure that best practices are being followed.
With Greenlight Guru Clinical, medical device companies can create customized electronic case report forms (eCRF) and digitize data collection for clinical investigations, in-human studies, and post-market surveillance activities.7
Post-market Surveillance Plan
What is it? The post-market surveillance (PMS) plan is part of a device’s required technical documentation, detailing your strategy for continuously monitoring and collecting data and safety information on the device. The plan is part of the requirements for a PMS system, and is intended to outline the criteria for the benefit-risk assessment of the device and processes for:
-
Collecting and analyzing data
-
Addressing submitted complaints
-
Communicating data to regulatory bodies and users
-
Implementing corrective actions on devices
Additionally, the PMS plan is used to determine whether or not a post-market clinical follow-up plan (PMCF) is required.
Where is it in the EU MDR? Article 84 requires that the PMS system is based on a plan. However, the details of the plan are specified in Annex III, 1.1.4
How to comply? A compliant PMS plan should consider information concerning serious incidents, records of non-serious incidents, available data on side-effects, information from trend reports, any feedback or complaints provided by users, distributors or importers of the medical device, and publicly available information about similar devices.
Annex III of EU MDR requires the following topics to be covered in the PMS plan:
-
A systematic and proactive process for collecting information (user feedback, reports, etc.)
-
Appropriate statistical and analytical methods of assessing collected data
-
Threshold values for assessing benefit-risk and effective risk management
-
Appropriate methods and tools for investigating complaints and other feedback collected in the field
-
Methods for managing events subject to the trend report
-
Protocols for communicating effectively with the competent authorities, notified bodies, economic operators, and users
-
Reference for procedures to fulfill the manufacturer's obligations for PMS system, PMS plan, and PSUR
-
Procedures and systems for implementing corrective actions when required
-
Effective tools to trace and identify devices for which corrective actions may be needed (traceability of potentially defective products in case a recall is needed, for example)
When do I submit or update the PMS plan? Your PMS plan must be established and documented prior to placing the medical device on the EU market for the first time, and updated as necessary during its lifecycle.
How can Greenlight Guru help you with your PMS plan? Post-market surveillance is a defining pillar of MDR. Throughout the regulation, we see many ways the PMS system touches or feeds into every part of the total product lifecycle, including the ongoing risk management and clinical evaluation processes.
If that wasn’t enough, the PMS plan must also address the processes used to collect and assess the massive amounts of data produced for a single device. This can be cause for concern among manufacturers, especially for those using a paper-based data collection process which carries high risk of missing and erroneous data.
Greenlight Guru Clinical makes it much more efficient to collect PMS data with direct data capture (DDC).6 Our electronic data capture (EDC) software lets medical device manufacturers seamlessly enter clinical data contemporaneously (during visits) or at a later point in time. Designed to fit MedTech needs, you don’t need technical skills to get started. Our ready-to-use templates, modules and features will help you to easily design the optimal clinical study.
Another benefit of our EDC software is that it allows clinicians to collect patient reported outcomes electronically on any device, thanks to bring-your-own-device (BYOD) capabilities. Our clients make full use of the ePRO capabilities by tailoring it to their clinical study needs as they solidify their device’s PMS plans.
Post-market Surveillance Report
What is it? A post-market surveillance report (PMSR) is a summary of the results and conclusions drawn from the data generated as part of the PMS plan. As part of a device’s technical documentation, it primarily serves as proof of your compliance with MDR requirements for post-market surveillance.
Post-market surveillance reports are used only for Class I devices, as opposed to the periodic safety update report required for higher-risk device classes.
Where is it in the EU MDR? Technical Documentation on Post Market Surveillance (Annex III 1.2.) or the equivalent for custom made devices (Annex XIII, Section 2).4
How to comply? Article 85 states that a PMSR must contain information summarizing the results and conclusions of the post-market data generated by the PMS plan. It also must be presented with a rationale and description for any preventive and corrective actions taken.
Additionally, while the PMSR is not required to be part of the feedback systems from the 8 or 9 processes laid out in Article 83, it may be beneficial to include status updates on each process for consistency’s sake.
When to submit or update a PMSR? The PMS report can be updated whenever the manufacturer considers it necessary and/or delivered upon request by an authority. But that means it must be produced/updated from time to time. At least once every three years is recommended.
How can Greenlight Guru support your Post-Market Surveillance Report? One of the most significant thematic changes in MDR is the requirement of manufacturers to now proactively gather input and data about device safety and clinical efficacy. Doing so requires your team to proactively collect information about safety issues, like adverse events (AEs) and severe adverse events (SAEs), which can be done by conducting customer surveys.
Depending on the PMS plan, surveys can be conducted with distributors, customers, and other actors that might have knowledge about safety issues or AEs. Surveys can then be scheduled out and performed routinely to identify any new events that originate in the market. This procedure for the constant monitoring of AEs should also be connected to your QMS, particularly for CAPA procedures..
With Greenlight Guru Quality, risk management activities can be linked to ensure full traceability. Additionally, Greenlight Guru Clinical was built with AE reporting modules that make it easy for clinical investigation teams to document, categorize, and report on AEs, SAEs, and medical device deficiencies in clinical investigations.6
While traditional EDC solutions require users to create a separate form or eCRF for reporting an AE/SAE, Greenlight Guru Clinical gives investigators access to ready-to-use AE form templates that reduce set-up time and eliminate duplicate documentation.
Post-market Clinical Follow-up Plan
What is it? The Post-market clinical follow-up plan (PMCF) specifies the methods and procedures used to proactively collect and evaluate clinical data on a device’s performance and safety. As part of your technical documentation, PMCF is connected to and used to update the PMS plan and CER. It also serves as the template for your PMCF report.
The goal of PMCF is to identify previously unknown side-effects, assess emergent risks, and prevent off-label misuse. This data can be captured through general PMCF activities, such as gathering feedback from end users and information from scientific literature.
However, in order to substantiate the data, it often is necessary to use specific PMCF activities—higher-level operations which can be used to scientifically illustrate safety and clinical performance, often based on case-specific data.10
These PMCF activities include:
-
PMCF studies (or observational or non-interventional clinical investigations)
-
Post-market interventional clinical investigations
-
Evaluation of data from suitable registries
-
Investigator initiated studies
-
Case cohorts
-
Other subject-specific clinical data collection activities
Where is it in the EU MDR? Post-Market Clinical Follow-Up activities are required by Part B of MDR’s Annex XIV, and further guidance and a template are available in the guidance document MDCG 2020-7.13
How to comply? The PMCF is a formal activity and cannot be performed in an ad hoc manner. You’ll need to create a post-market clinical follow-up plan that outlines the process you intend to use to gather and evaluate data for your PMCF evaluation report.
Generally speaking, your PMCF plan will have seven sections:
-
The manufacturer’s contact details
-
A description and specification of the medical device being studied
-
The activities related to the PCMF (these are both the general and specific methods and procedures you’re using)
-
References to any relevant parts of the technical documentation
-
An evaluation of clinical data for equivalent or similar devices
-
References to any applicable common specifications, harmonized standards, or guidance documents
-
Estimated date of the PMCF evaluation report
When to submit or update the PMCF plan? By definition, post-market clinical follow-up activities occur after a medical device has been placed on the market. However, PMCF plans are part of the PMS system, and should there be any corrective actions taken as a result of the clinical studies, the PMCF plan should be updated to reflect those changes.
How can Greenlight Guru help you with your PMCF plan? A common pitfall for medical device companies planning their PMCF activities are communication breakdowns, such as the need for approval of ethical committees, as well as data silos that prevent key stakeholder opinions from staying connected. PMCF planning is a time-consuming task and the increased cost from this step is often a surprise for medical devices companies with limited PMCF experience.
With Greenlight Guru Clinical, you can use specific templates engineered for medical device data collection, reusable across products and markets. Your team can make generic product-related questions for safety and performance. Also, you can select specific scales to track both.
You can also benefit from the centralization of both your clinical data and your quality management system data, thanks to Greenlight Guru’s industry-leading eQMS solution. Linking all these items in a single workflow environment means you won’t be looking for missing documentation, lost signatures, or any of the other frustrations that come with outdated paper-based tracking.
Post-market Clinical Follow-up Report
What is it? The Post-market Clinical Follow-up Report is the structured summarization of the resulting data from the PMCF plan. Recording and reporting of adverse events and serious adverse events is a key aspect of a successful clinical investigation and staying on top of regulatory changes is a must.
Where is it in the EU MDR? PMCF reports are required under Annex XIV Part B of the MDR.2 Further guidance can be found in MDCG 2020-8, which outlines the reporting requirements and provides a template.14
How to comply? PMCF activities conducted for your device must follow the latest requirements for GCP (Good Clinical Practice).16 As part of GCP, you are required to notify competent authorities of serious incidents that were previously left undocumented, such as previously unknown side-effects. Additionally, you’ll need to clearly describe this incident in the product documentation prior to introducing your device into the EU market.
When to submit or update? Just like PMCF plans, the PMCF reports will naturally come during your post-market surveillance phase, and will subsequently require being updated whenever appropriate.
How can Greenlight Guru help you with your PMCF report? One of the critical quality requirements for both clinical investigations and PMCF under the MDR is compliance with GCP. To ensure that the data collected in a PMCF survey complies with the quality standards of the EU MDR, sponsors have to use solutions that can fulfill the ISO 14155:2020 requirements for electronic data capture.17
This means that if you use survey software that does not comply with ISO 14155:2020, you risk not being able to use the PMCF survey data in your clinical evaluation report. Which then can potentially lead to a loss of CE mark.
To avoid this, you’ll need to ensure your EDC software provider can:
-
Provide documentation of verification and validation according to the requirements specified in ISO 14155:2020
-
Document data traceability from source to sponsor
-
Comply with all standard GCP principles when collecting, viewing, editing, and exporting data
-
Store the data according to industry standards on general security and permission control
-
Comply with GDPR requirements for informed consents and data protection
With Greenlight Guru Clinical, you can rest assured knowing that our EDC solution is pre-validated and compliant with both ISO 14155:2020 and GCP. It’s also why our medical device-specific solution is designed to stay up-to-date with the latest FDA, ISO, and EU MDR guidelines for post-market and clinical study needs.
![eu-device-requirements-class-IIa]() Device Class IIa Requirements
Device Class IIa Requirements
 Device Class IIa Requirements
Device Class IIa RequirementsClass IIa medical devices are considered medium-risk devices by the MDR. This means that unlike a Class I device, the manufacturer must receive a declaration of conformity from a notified body following its conformity assessment. Examples of this risk class include catheters, hearing aids, or short-term contact lenses.
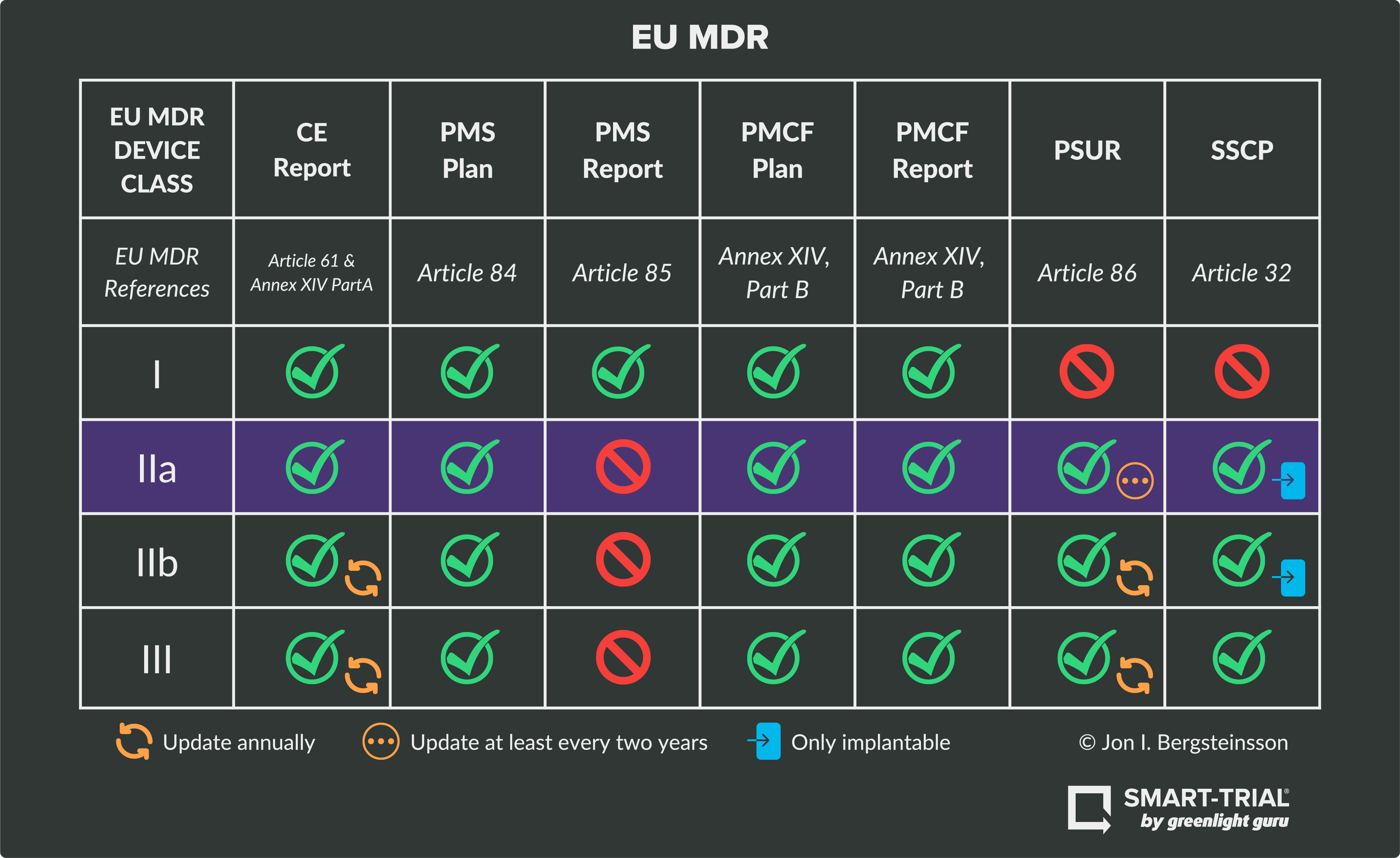
As seen in the graphic above, Class IIa devices must fulfill obligations and technical requirements for:
-
Clinical Evaluation Report (CER)
-
Post-market Surveillance Plan (PMS Plan)
-
Periodic Safety Update Report (PSUR)
-
Post-market Clinical Follow-up Plan (PMCF Plan)
-
Post-market Clinical Follow-up Report (PMCF Report)
-
Summary of Safety and Clinical Performance (SSCP)
NOTE: Class IIa devices do not require a PMS report.
CE Report
What is it? The clinical evaluation report (CER) is documentation of the clinical evaluation that is required of every medical device sold in the EU. Its purpose is to prove your device performs as intended without compromising the safety of its end users.
How is it regulated under EU MDR? Article 61 of EU MDR requires every medical device manufacturer to document the clinical evaluation of their device in a CER.3 This requirement is expanded upon in Annex XIV Part A, which states:
The results of the clinical evaluation and the clinical evidence on which it is based shall be documented in a clinical evaluation report which shall support the assessment of the conformity of the device.
MDCG-2020-5 and MDCG 2020-13 are guidance documents for clinical evaluations, and contain instructions and specific requirements for the clinical evaluation report.5
How to comply? MDCG-2020-5 states that your CER should contain sufficient information for anyone reading it to understand the search criteria, the included data, and all assumptions made as well as all conclusions reached.
MDCG-2020-13 provides a sample table of contents, which indicate the CER should include:
-
Administrative particulars
-
Medical device name model and type
-
Manufacturer(s) name and SRN
-
Notified body
-
Type of assessment
-
Intended purpose
-
Check of clinical evaluation report authors
-
Reviewers involved in notified body assessment of the clinical evaluation
-
Device information, including:
-
Description
-
Classification
-
Clinical evaluation plan
-
Information materials supplied by the manufacturer
-
Common specifications and harmonised standards applied
-
Demonstration of equivalence
-
State of the art
-
Clinical literature review
-
Clinical investigations and related documentation
-
PMS, PMCF and the plan for updates
-
IFU, SSCP, labeling and other information supplied with the device
-
Summary of all available data and conclusions
-
Overall Conclusions
Lastly, the guidance states that the report’s contents shall be cross-referenced to supporting documents, and to their locations in your technical documentation.
When do I submit or update the CE Report? The CER is meant to be a living document, and if at any point you receive new and pertinent information about your device through postmarket surveillance or new clinical evaluations, then the CER must be updated to reflect that. However, even if you don’t receive new information, your clinical evaluation report must still be updated according to a schedule that you, the manufacturer, define and justify.
For devices without significant risk, the CER must be updated every two to five years. The schedule you choose should reflect the risk classification of your device and how well-established the technology behind the device is.
How can Greenlight Guru help you manage the entire clinical evaluation process? A successful clinical evaluation report hinges on the quality of clinical data and the processes for collecting and analyzing it. To do so, researchers use Case Report Forms (CRFs) completed by investigators for each patient during the investigation.8
In the past, CRFs from a clinical study would be completed on paper, packed into boxes, and shipped from the study location to a secondary location for analysis. But today, a growing number of medical device companies are going paperless and using web-based Electronic Data Capture (EDC) systems to collect, store, and manage clinical data.9
Software-based EDC systems give clinical study managers the ability to streamline the data collection process, capture more accurate data, and enhance data security and accessibility - all while saving time and reducing the cost of completing a clinical study. EDC systems are also an essential pairing with a well-developed quality management system to ensure that best practices are being followed.
With Greenlight Guru Clinical, medical device companies can create customized electronic case report forms (eCRF) and digitize data collection for clinical investigations, in-human studies, and post-market surveillance activities.7
Post-market Surveillance Plan
What is it? The post-market surveillance (PMS) plan is part of a device’s required technical documentation, detailing your strategy for continuously monitoring and collecting data and safety information on the device. The plan is part of the requirements for a PMS system, and is intended to outline the criteria for the benefit-risk assessment of the device and processes for:
-
Collecting and analyzing data
-
Addressing submitted complaints
-
Communicating data to regulatory bodies and users
-
Implementing corrective actions on devices
Additionally, the PMS plan is used to determine whether or not a post-market clinical follow-up plan (PMCF) is required.
Where is it in the EU MDR? Article 84 requires that the PMS system is based on a plan.4 However, the details of the plan are specified in Annex III, 1.1.
How to comply? A compliant PMS plan should consider information concerning serious incidents, records of non-serious incidents, available data on side-effects, information from trend reports, any feedback or complaints provided by users, distributors or importers of the medical device, and publicly available information about similar devices.
Annex III of EU MDR requires the following topics to be covered in the PMS plan:
-
A systematic and proactive process for collecting information (user feedback, reports, etc.)
-
Appropriate statistical and analytical methods of assessing collected data
-
Threshold values for assessing benefit-risk and effective risk management
-
Appropriate methods and tools for investigating complaints and other feedback collected in the field
-
Methods for managing events subject to the trend report
-
Protocols for communicating effectively with the competent authorities, notified bodies, economic operators, and users
-
Reference for procedures to fulfill the manufacturer's obligations for PMS system, PMS plan, and PSUR
-
Procedures and systems for implementing corrective actions when required
-
Effective tools to trace and identify devices for which corrective actions may be needed (traceability of potentially defective products in case a recall is needed, for example)
When do I submit or update the PMS plan? The PMS plan must be established and documented prior to placing the medical device on the EU market for the first time, and updated as necessary during its lifecycle.
How can Greenlight Guru help you with your PMS plan? Post-market surveillance is a defining pillar of MDR. Throughout the regulation, we see many ways the PMS system touches or feeds into every part of the total product lifecycle, including the ongoing risk management and clinical evaluation processes.
If that wasn’t enough, the PMS plan must also address the processes used to collect and assess the massive amounts of data produced for a single device. This can be cause for concern among manufacturers, especially for those using a paper-based data collection process which carries high risk of missing and erroneous data.
Greenlight Guru Clinical makes it much more efficient to collect PMS data with direct data capture (DDC).6 Our electronic data capture (EDC) software lets medical device manufacturers seamlessly enter clinical data contemporaneously (during visits) or at a later point in time. Designed to fit MedTech needs, you don’t need technical skills to get started. Our ready-to-use templates, modules and features will help you to easily design the optimal clinical study.
Another benefit of our EDC software is that it allows clinicians to collect patient reported outcomes electronically on any device, thanks to bring-your-own-device (BYOD) capabilities. Our clients make full use of the ePRO capabilities by tailoring it to their clinical study needs as they solidify their device’s PMS plans.
Periodic Safety Update Report
What is it? The periodic safety update report is a summary of the results of postmarket surveillance activities as well as the conclusions that manufacturers have drawn from those results. If the manufacturer has taken any corrective or preventive actions (CAPAs), a description and rationale for the actions must also be included in this report.
The periodic safety update report is part of a device’s technical documentation, and it has to be updated throughout the device’s lifecycle. The PSUR is initially submitted to a Notified Body during the device’s conformity assessment audit, but from then on it must be updated either annually or biennially.
Where is it in the EU MDR? The full requirements for your PSUR can be found in Article 86 of MDR,11 or Article 81 of IVDR.12
How to comply? PSURs are identical to PMSRs with a few additions - manufacturers must publish the conclusion of the benefit-risk determination, main findings of the post-market clinical or performance follow-up, sales volume, and estimated user population characteristics and usage frequency.
When to submit or update? Manufacturers of Class IIa devices shall update the PSUR when necessary and at least every two years. The PSUR shall, except in the case of custom-made devices, be part of the technical documentation as specified in Annexes II and III.
How can Greenlight Guru support your PSUR? The periodic safety update report is not just a data dump or a chance to check some boxes. It is intended to be an analysis of the post-market data your company has actively collected. It’s a meaningful, regularly updated report that should help both regulators and internal stakeholders understand the safety and efficacy of your device during its time on the market.
In order to prove your marketed device is safe, effective, and successful at fulfilling its intended purpose, you need to provide rationale in the form of data-driven and meaningful conclusions as a result of your PMS activities and clinical follow-ups. This can be difficult for companies — what if your argument doesn’t work? What if your conclusions are unconvincing, or customer data on usage are spotty?
This concern demonstrates the need for using validated questionnaires, which are especially useful when you need to gather data for regulatory purposes since they are scientifically tested. The results from these questionnaires can provide reliable information on the topic of interest - if applied correctly. In addition, it also simplifies the justification of your data collection method, to why you are asking specific questions.
With Greenlight Guru Clinical, creating custom questionnaires and forms to use in a PMCF survey or clinical investigational context often returns a more complex dataset. Standardized questionnaires are helpful because they provide sponsors with a structured dataset that is easy to analyze. It minimizes the need to clean data or merge open-ended or text-based answers. In the end it can save a great amount of time in creating an accurate and thorough PSUR.
Post-market Clinical Follow-up Plan
What is it? The post-market clinical follow-up plan (PMCF) specifies the methods and procedures used to proactively collect and evaluate clinical data on a device’s performance and safety. As part of your technical documentation, PMCF is connected to and used to update your PMS plan and CER. It also serves as the template for your PMCF report.
The goal of PMCF is to identify previously unknown side-effects, assess emergent risks, and prevent off-label misuse. This data can be captured through general PMCF activities, such as gathering feedback from end users and information from scientific literature.
However, in order to substantiate the data, it often is necessary to use specific PMCF activities—higher-level operations which can be used to scientifically illustrate safety and clinical performance, often based on case-specific data.
These PMCF activities include:
-
PMCF studies (or observational or non-interventional clinical investigations)
-
Post-market Interventional clinical investigations
-
Evaluation of data from suitable registries
-
Investigator initiated studies
-
Case cohorts
-
Other subject-specific clinical data collection activities
Where is it in the EU MDR? Post-Market Clinical Follow-Up activities are required by Part B of MDR’s Annex XIV, and further guidance and a template are available in the guidance document MDCG 2020-7.13
How to comply? The PMCF is a formal activity and cannot be performed in an ad hoc manner. You’ll need to create a post-market clinical follow-up plan that outlines the process you intend to use to gather and evaluate data for your PMCF evaluation report.
Generally speaking, your PMCF plan will have seven sections:
-
The manufacturer’s contact details
-
A description and specification of the medical device being studied
-
The activities related to the PCMF (these are both the general and specific methods and procedures you’re using)
-
References to any relevant parts of the technical documentation
-
An evaluation of clinical data for equivalent or similar devices
-
References to any applicable common specifications, harmonized standards, or guidance documents
-
Estimated date of the PMCF evaluation report
When to submit or update the PMCF plan? By definition, Post-Market Clinical Follow-up activities occur after a medical device has been placed on the market. However, PMCF plans are part of the PMS system, and should there be any corrective actions taken as a result of the clinical studies, the PMCF should be updated to reflect those changes.
How can Greenlight Guru help you with your PMCF plan? A common pitfall for medical device companies planning their PMCF activities are communication breakdowns, such as the need for approval of ethical committees, as well as data silos that prevent key stakeholder opinions from staying connected. PMCF planning is a time-consuming task and the increased cost from this step is often a surprise for medical devices companies with limited PMCF experience.
With Greenlight Guru Clinical, you can use a personalized PMCF plan template that is reusable across products and markets. Your team can make generic product-related questions for safety and performance. Also, you can select specific scales to track both.
You can also benefit from the centralization of both your clinical data and your quality management system data, thanks to Greenlight Guru’s industry-leading eQMS solution. Linking all these items in a single workflow environment means you won’t be looking for missing documentation, lost signatures, or any of the other frustrations that come with outdated paper-based tracking.
Post-market Clinical Follow-up Report
What is it? The Post-market Clinical Follow-up Report is the structured summarization of the resulting data from the PMCF plan. Recording and reporting of adverse events and serious adverse events is a key aspect of a successful clinical investigation and staying on top of regulatory changes is a must.
Where is it in the EU MDR? PMCF reports are required under Annex XIV Part B of the MDR.2 Further guidance can be found in MDCG 2020-8, which outlines the reporting requirements and provides a template.14
How to comply? PMCF activities conducted for your device must follow the latest requirements for GCP (Good Clinical Practice).16 As part of GCP, you are required to notify competent authorities of serious incidents that were previously left undocumented, such as previously unknown side-effects. Additionally, you’ll need to clearly describe this incident in the product documentation prior to introducing your device into the EU market.
When to submit or update? Just like PMCF plans, the PMCF reports will naturally come during your post-market surveillance phase, and will subsequently require being updated whenever appropriate.
How can Greenlight Guru help you with your PMCF report? One of the critical quality requirements for both clinical investigations and PMCF under the MDR is compliance with GCP. To ensure that the data collected in a PMCF survey complies with the quality standards of the EU MDR, sponsors have to use solutions that can fulfill the ISO 14155:2020 requirements for electronic data capture.17
This means that if you use survey software that does not comply with ISO 14155:2020, you risk not being able to use the PMCF survey data in your clinical evaluation report. Which then can potentially lead to a loss of CE mark.
To avoid this, you’ll need to ensure your EDC software provider can:
-
Provide documentation of verification and validation according to the requirements specified in ISO 14155:2020
-
Document data traceability from source to sponsor
-
Comply with all standard GCP principles when collecting, viewing, editing, and exporting data
-
Store the data according to industry standards on general security and permission control
-
Comply with GDPR requirements for informed consents and data protection
With Greenlight Guru Clinical, you can rest assured knowing that our EDC solution is pre-validated and compliant with both ISO 14155:2020 and GCP. It’s also why our medical device-specific solution is designed to stay up-to-date with the latest FDA, ISO, and EU MDR guidelines for post-market and clinical study needs.
Summary of Safety and Clinical Performance
What is it? The summary of safety and clinical performance is an external document that incorporates information related to a medical device, such as general information, a summary of the clinical data collected from the device, or possible therapeutic alternatives.
SSCP is intended for devices which are higher risk or invasive, specifically:
-
Implantable Class IIa devices
-
Implantable Class IIb devices
-
All Class III devices
The main purpose of the SSCP is to provide both healthcare workers and patients access to current clinical, safety, performance, and other types of data from the medical device; as this aligns with the MDR philosophy of providing a more transparent and robust regulatory framework.
Where is it in the EU MDR? The requirements and general definition for SSCP/SSP can be found in MDR, under Article 32. For further instruction, consult the guidance document MDCG 2019-9 – Rev.1.15
How to comply? The SSCP/SSP must be objective, including both favorable and unfavorable data. It should be based on the device's technical documentation and constantly revised to prevent outdated information.
MDCG 2019-9 – Rev.1 states the SSCP must at minimum contain sections on:
-
Device and manufacturer general details – including UDI details
-
Intended purpose
-
Indications, contraindications and target population
-
Description of the device, its components, medicinal products and any previous version of the device
-
Residual risk, warning and precautions details
-
Summary of the Clinical Evaluation of the device, emphasizing the PMCF evaluation and pointing out conclusions related to safety and performance
-
Description of necessary training and qualifications
-
Reference to harmonized standards and common specifications applied
-
Revision history
When to submit or update? Implantable Class IIa devices must prepare SSCPs annually, and may include updates from the Post-Market Clinical Follow-up (PMCF) and the Product Safety Update Report (PSUR) as part of its ongoing lifecycle.
How can Greenlight Guru support your SSCP? The SSCP needs to translate technical documentation into messaging that anyone can understand, not just those trained to be medical device experts.
This document will be a guide for providers and patients alike, and therefore can’t be viewed as a piece of marketing material. The information must be objective, sometimes even negative, while still balancing transparency with the needs for user privacy. In short—writing these documents is an art.
The solution for this problem is to simplify the process with templates that work well, together. With Greenlight Guru Clinical, we deliver out-of-the-box compliance with ISO 14155:2020, along with user-friendly eCRF design templates, GDPR-compliant data export, and support for many different study designs and protocols.
![eu-device-requirements-class-IIb]() Device Class IIb Requirements
Device Class IIb Requirements
 Device Class IIb Requirements
Device Class IIb RequirementsClass IIb medical devices are considered medium- to high-risk devices under the MDR, and thus their CE route also requires the involvement of a notified body. This risk class includes devices like incubators, insulin pens, long-term contact lenses, and ventilators.
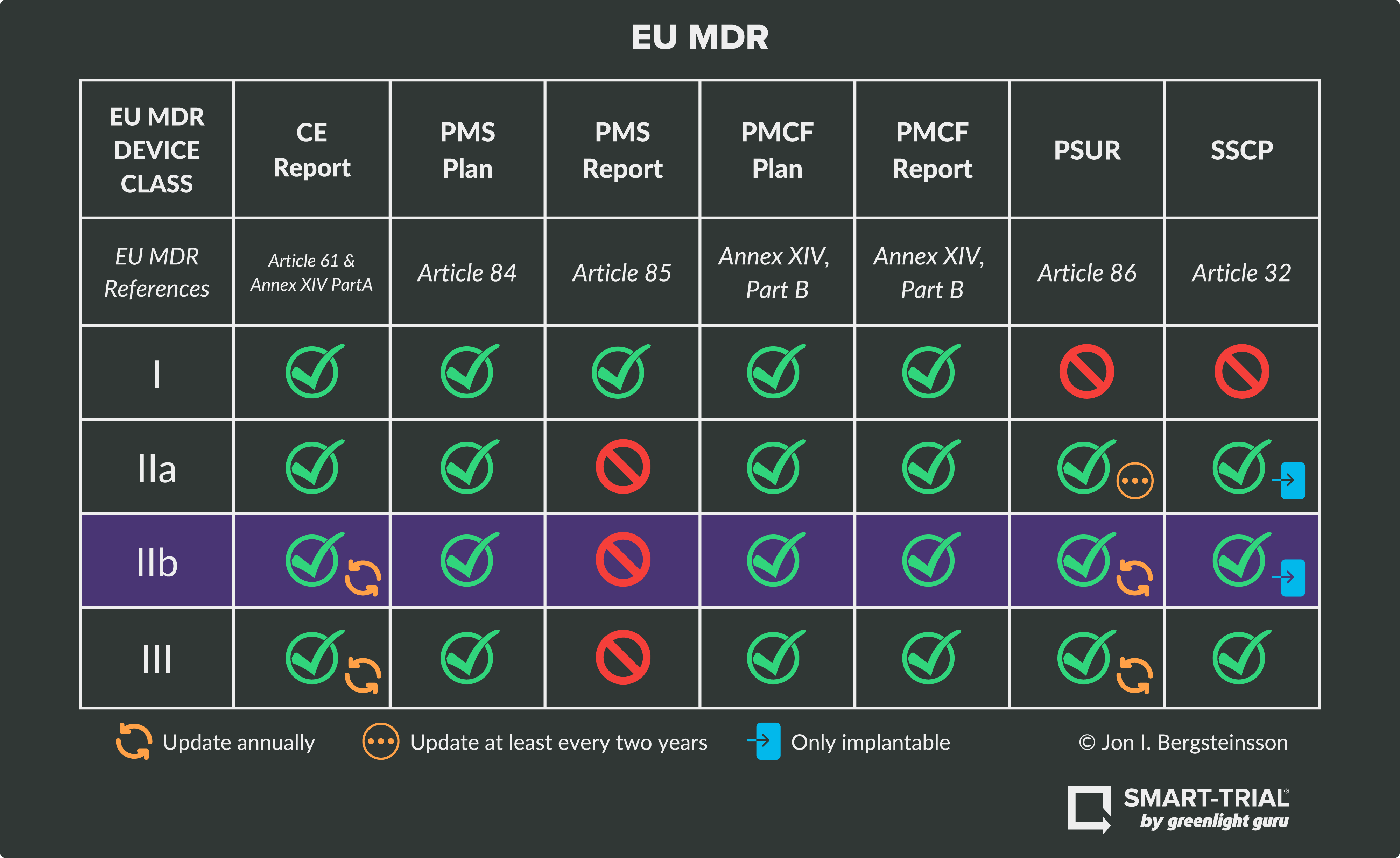
As seen in the graphic above, Class IIb devices must fulfill obligations and technical requirements for:
- Clinical Evaluation Report (CER)
- Post-market Surveillance Plan (PMS Plan)
- Periodic Safety Update Report (PSUR)
- Post-market Clinical Follow-up Plan (PMCF Plan)
- Post-market Clinical Follow-up Report (PMCF Report)
- Summary of Safety and Clinical Performance (SSCP)
NOTE: Class IIb devices DO NOT require a PMS report.
Without further ado, let’s jump into the specifics of each safety and technical requirement for your Class IIb device under EU MDR.
CE Report
What is it? The clinical evaluation report (CER) is documentation of the clinical evaluation that is required of every medical device sold in the EU. Its purpose is to prove your device performs as intended without compromising the safety of its end users.
How is it regulated under EU MDR? Article 61 of EU MDR requires every medical device manufacturer to document the clinical evaluation of their device in a CER.3 This requirement is expanded upon in Annex XIV Part A, which states:
The results of the clinical evaluation and the clinical evidence on which it is based shall be documented in a clinical evaluation report which shall support the assessment of the conformity of the device.
MDCG-2020-5 and MDCG 2020-13 are guidance documents for clinical evaluations, and contain instructions and specific requirements for the clinical evaluation report.5
How to comply? MDCG-2020-5 states that your CER should contain sufficient information for anyone reading it to understand the search criteria, the included data, and all assumptions made as well as all conclusions reached.
MDCG-2020-13 provides a sample table of contents, which indicate the CER should include:
-
Administrative particulars
-
Medical device name model and type
-
Manufacturer(s) name and SRN
-
Notified body
-
Type of assessment
-
Intended purpose
-
Check of clinical evaluation report authors
-
Reviewers involved in notified body assessment of the clinical evaluation
-
Device information, including:
-
Description
-
Classification
-
Clinical evaluation plan
-
Information materials supplied by the manufacturer
-
Common specifications and harmonised standards applied
-
Demonstration of equivalence
-
State of the art
-
Clinical literature review
-
Clinical investigations and related documentation
-
PMS, PMCF and the plan for updates
-
IFU, SSCP, labeling and other information supplied with the device
-
Summary of all available data and conclusions
-
Overall Conclusions
Lastly, the guidance states that the report’s contents shall be cross-referenced to supporting documents, and to their locations in your technical documentation.
When do I submit or update the CE Report? The CE report for Class IIb devices is intended to be a living document, and therefore must be updated annually, as well as at any other time it is relevant to do so, such as when significant changes occur.
How can Greenlight Guru help you manage the entire clinical evaluation process? A successful clinical evaluation report hinges on the quality of clinical data and the processes for collecting and analyzing it. To do so, researchers use Case Report Forms (CRFs) completed by investigators for each patient during the investigation.8
In the past, CRFs from a clinical study would be completed on paper, packed into boxes, and shipped from the study location to a secondary location for analysis. But today, a growing number of medical device companies are going paperless and using web-based Electronic Data Capture (EDC) systems to collect, store, and manage clinical data.9
Software-based EDC systems give clinical study managers the ability to streamline the data collection process, capture more accurate data, and enhance data security and accessibility - all while saving time and reducing the cost of completing a clinical study. EDC systems are also an essential pairing with a well-developed quality management system to ensure that best practices are being followed.
With Greenlight Guru Clinical, medical device companies can create customized electronic case report forms (eCRF) and digitize data collection for clinical investigations, in-human studies, and post-market surveillance activities.7
Post-market Surveillance Plan
What is it? The post-market surveillance (PMS) plan is part of a device’s required technical documentation, detailing your strategy for continuously monitoring and collecting data and safety information on the device. The plan is part of the requirements for a PMS system, and is intended to outline the criteria for the benefit-risk assessment of the device and processes for:
-
Collecting and analyzing data
-
Addressing submitted complaints
-
Communicating data to regulatory bodies and users
-
Implementing corrective actions on devices
Additionally, the PMS plan is used to determine whether or not a post-market clinical follow-up plan (PMCF) is required.
Where is it in the EU MDR? Article 84 requires that the PMS system is based on a plan.4 However, the details of the plan are specified in Annex III, 1.1.
How to comply? A compliant PMS plan should consider information concerning serious incidents, records of non-serious incidents, available data on side-effects, information from trend reports, any feedback or complaints provided by users, distributors or importers of the medical device, and publicly available information about similar devices.
Annex III of EU MDR requires the following topics to be covered in the PMS plan:
-
A systematic and proactive process for collecting information (user feedback, reports, etc.)
-
Appropriate statistical and analytical methods of assessing collected data
-
Threshold values for assessing benefit-risk and effective risk management
-
Appropriate methods and tools for investigating complaints and other feedback collected in the field
-
Methods for managing events subject to the trend report
-
Protocols for communicating effectively with the competent authorities, notified bodies, economic operators, and users
-
Reference for procedures to fulfill the manufacturer's obligations for PMS system, PMS plan, and PSUR
-
Procedures and systems for implementing corrective actions when required
-
Effective tools to trace and identify devices for which corrective actions may be needed (traceability of potentially defective products in case a recall is needed, for example)
When do I submit or update the PMS plan? The PMS plan must be established and documented prior to placing the medical device on the market for the first time, and updated as necessary during its lifecycle.
How can Greenlight Guru help you with your PMS plan? Post-market surveillance is a defining pillar of MDR. Throughout the regulation, we see many ways the PMS system touches or feeds into every part of the total product lifecycle, including the ongoing risk management and clinical evaluation processes.
If that wasn’t enough, the PMS plan must also address the processes used to collect and assess the massive amounts of data produced for a single device. This can be cause for concern among manufacturers, especially for those using a paper-based data collection process which carries high risk of missing and erroneous data.
Greenlight Guru Clinical makes it much more efficient to collect PMS data with direct data capture (DDC).6 Our electronic data capture (EDC) software lets medical device manufacturers seamlessly enter clinical data contemporaneously (during visits) or at a later point in time. Designed to fit MedTech needs, you don’t need technical skills to get started. Our ready-to-use templates, modules and features will help you to easily design the optimal clinical study.
Another benefit of our EDC software is that it allows clinicians to collect patient reported outcomes electronically on any device, thanks to bring-your-own-device (BYOD) capabilities. Our clients make full use of the ePRO capabilities by tailoring it to their clinical study needs as they solidify their device’s PMS plans.
Periodic Safety Update Report
What is it? The Periodic Safety Update Report is a summary of the results of postmarket surveillance activities as well as the conclusions that manufacturers have drawn from those results. If the manufacturer has taken any corrective or preventive actions (CAPAs), a description and rationale for the actions must also be included in this report.
The Periodic Safety Update Report is part of a device’s technical documentation, and it has to be updated throughout the device’s lifecycle. The PSUR is initially submitted to a notified body during the device’s conformity assessment audit, but from then on it must be updated either annually or biennially.
Where is it in the EU MDR? The full requirements for your PSUR can be found in Article 86 of MDR,11 or Article 81 of IVDR.12
How to comply? PSURs are identical to PMSRs with a few additions - manufacturers must publish the conclusion of the benefit-risk determination, main findings of the post-market clinical or performance follow-up, sales volume, and estimated user population characteristics and usage frequency.
When to submit or update? Manufacturers of Class IIb devices shall update the PSUR when necessary and at least every two years. That PSUR shall, except in the case of custom-made devices, be part of the technical documentation as specified in Annexes II and III.
How can Greenlight Guru support your PSUR? The Periodic Safety Update Report is not just a data dump or a chance to check some boxes. It is intended to be an analysis of the postmarket data your company has actively collected. It’s a meaningful, regularly updated report that should help both regulators and internal stakeholders understand the safety and efficacy of your device during its time on the market.
In order to help readers reach the level of understanding—that this marketed device is safe, effective, and successful at fulfilling its intended purpose—you need to provide rationale in the form of data-driven and meaningful conclusions as a result of your postmarket surveillance and clinical followups. This can be difficult for companies — what if your argument doesn’t work? What if your conclusions are unconvincing, or customer data on usage are spotty?
This concern demonstrates the need for using validated questionnaires. Validated questionnaires are especially useful when you need to gather data for regulatory purposes because they are scientifically tested. This means that the results from those questionnaires can provide reliable information on the topic of interest - if applied correctly. In addition, it also simplifies the justification of your data collection method, to why you are asking specific questions.
With Greenlight Guru Clinical, creating custom questionnaires and forms to use in a PMCF survey or clinical investigational context often returns a more complex dataset. Standardized questionnaires are helpful, because they provide sponsors with a structured dataset that is easy to analyze. It minimizes the need to clean data or merge open-ended or text-based answers. In the end it can save a great amount of time in creating a competent and thorough PSUR.
Post-market Clinical Follow-up Plan
What is it? The Post-market clinical follow-up plan (PMCF) specifies the methods and procedures used to proactively collect and evaluate clinical data on a device’s performance and safety. As part of your technical documentation, PMCF is connected to and used to update the PMS plan and CER. It also serves as the template for your PMCF report.
The goal of PMCF is to identify previously unknown side-effects, assess emergent risks, and prevent off-label misuse. This data can be captured through general PMCF activities, such as gathering feedback from end users and information from scientific literature.
However, in order to substantiate the data, it often is necessary to use specific PMCF activities—higher-level operations which can be used to scientifically illustrate safety and clinical performance, often based on case-specific data.10
These PMCF activities include:
-
PMCF studies (or observational or non-interventional clinical investigations)
-
Post-market interventional clinical investigations
-
Evaluation of data from suitable registries
-
Investigator initiated studies
-
Case cohorts
-
Other subject-specific clinical data collection activities
Where is it in the EU MDR? Post-Market Clinical Follow-Up activities are required by Part B of MDR’s Annex XIV, and further guidance and a template are available in the guidance document MDCG 2020-7.13
How to comply? The PMCF is a formal activity and cannot be performed in an ad hoc manner. You’ll need to create a post-market clinical follow-up plan that outlines the process you intend to use to gather and evaluate data for your PMCF evaluation report.
Generally speaking, your PMCF plan will have seven sections:
-
The manufacturer’s contact details
-
A description and specification of the medical device being studied
-
The activities related to the PCMF (these are both the general and specific methods and procedures you’re using)
-
References to any relevant parts of the technical documentation
-
An evaluation of clinical data for equivalent or similar devices
-
References to any applicable common specifications, harmonized standards, or guidance documents
-
Estimated date of the PMCF evaluation report
When to submit or update the PMCF plan? By definition, Post-Market Clinical Follow-up activities occur after a medical device has been placed on the market. However, PMCF plans are part of the PMS system, and should there be any corrective actions taken as a result of the clinical studies, the PMCF should be updated to reflect those changes.
How can Greenlight Guru help you with your PMCF plan? A common pitfall for medical device companies planning their PMCF activities are communication breakdowns, such as the need for approval of ethical committees, as well as data silos that prevent key stakeholder opinions from staying connected. PMCF planning is a time-consuming task and the increased cost from this step is often a surprise for medical devices companies with limited PMCF experience.
With Greenlight Guru Clinical, you can use a personalized PMCF plan template that is reusable across products and markets. Your team can make generic product-related questions for safety and performance. Also, you can select specific scales to track both.
You can also benefit from the centralization of both your clinical data and your quality management system data, thanks to Greenlight Guru’s industry-leading eQMS solution. Linking all these items in a single workflow environment means you won’t be looking for missing documentation, lost signatures, or any of the other frustrations that come with outdated paper-based tracking.
Post-market Clinical Follow-up Report
What is it? The Post-market Clinical Follow-up Report is the structured summarization of the resulting data from the PMCF plan. Recording and reporting of adverse events and serious adverse events is a key aspect of a successful clinical investigation and staying on top of regulatory changes is a must.
Where is it in the EU MDR? PMCF reports are required under Annex XIV Part B of the MDR.2 Further guidance can be found in MDCG 2020-8, which outlines the reporting requirements and provides a template.14
How to comply? PMCF activities conducted for your device must follow the latest requirements for GCP (Good Clinical Practice).16 As part of GCP, you are required to notify competent authorities of serious incidents that were previously left undocumented, such as previously unknown side-effects. Additionally, you’ll need to clearly describe this incident in the product documentation prior to introducing your device into the EU market.
When to submit or update? Just like PMCF plans, the PMCF reports will naturally come during your post-market surveillance phase, and will subsequently require being updated whenever appropriate.
How can Greenlight Guru help you with your PMCF report? One of the critical quality requirements for both clinical investigations and PMCF under the MDR is compliance with GCP. To ensure that the data collected in a PMCF survey complies with the quality standards of the EU MDR, sponsors have to use solutions that can fulfill the ISO 14155:2020 requirements for electronic data capture.17
This means that if you use survey software that does not comply with ISO 14155:2020, you risk not being able to use the PMCF survey data in your clinical evaluation report. Which then can potentially lead to a loss of CE mark.
To avoid this, you’ll need to ensure your EDC software provider can:
-
Provide documentation of verification and validation according to the requirements specified in ISO 14155:2020
-
Document data traceability from source to sponsor
-
Comply with all standard GCP principles when collecting, viewing, editing, and exporting data
-
Store the data according to industry standards on general security and permission control
-
Comply with GDPR requirements for informed consents and data protection
With Greenlight Guru Clinical, you can rest assured knowing that our EDC solution is pre-validated and compliant with both ISO 14155:2020 and GCP. It’s also why our medical device-specific solution is designed to stay up-to-date with the latest FDA, ISO, and EU MDR guidelines for post-market and clinical study needs.
Summary of Safety and Clinical Performance
What is it? An SSCP/SSP is an external document that incorporates information related to a medical device, such as general information, a summary of the clinical data collected from the device, or possible therapeutic alternatives. SSCP is intended for devices which are higher risk or invasive, specifically:
-
Implantable Class IIa devices
-
Implantable Class IIb devices
-
All Class III devices
The main purpose of the SSCP/SSP is to provide both healthcare workers and patients access to current clinical, safety, performance and other types of data from the medical device; as this aligns with the MDR/IVDR philosophy of providing a more transparent and robust regulatory framework.
Where is it in the EU MDR? The requirement and general definition is found in MDR, under Article 32. For further instruction, consult the guidance doc MDCG 2019-9 – Rev.1.15
How to comply? The SSCP/SSP must be objective, including both favorable and unfavorable data. It should be based on the device's technical documentation and constantly revised to prevent outdated information.
MDCG 2019-9 – Rev.1 states the SSCP must at minimum contain sections on:
-
Device and manufacturer general details – including UDI details
-
Intended purpose
-
Indications, contraindications and target population
-
Description of the device, its components, medicinal products and any previous version of the device
-
Residual risk, warning and precautions details
-
Summary of the clinical evaluation of the device, emphasizing the PMCF evaluation and pointing out conclusions related to safety and performance
-
Description of necessary training and qualifications
-
Reference to harmonized standards and common specifications applied
-
Revision history
When to submit or update? Implantable Class IIb devices must prepare SSCPs annually, and may include updates from the Post-Market Clinical Follow-up (PMCF) and the Product Safety Update Report (PSUR) as part of its ongoing lifecycle.
How can Greenlight Guru support your SSCP? The SSCP needs to translate technical documentation into messaging that anyone can understand, not just those trained to be medical device experts.
This document will be a guide for providers and patients alike, and therefore can’t be viewed as a piece of marketing material. The information must be objective, sometimes even negative, while still balancing transparency with the needs for user privacy. In short—writing these documents is an art.
The solution for this problem is to simplify the process with templates that work well, together. With Greenlight Guru Clinical, we deliver out-of-the-box compliance with ISO 14155:2020, along with user-friendly eCRF design templates, GDPR-compliant data export, and support for many different study designs and protocols.
And because the SSCP plays so heavily into ISO requirements for continuous improvement and quality management systems, our EDC software pairs seamlessly with our eQMS solution and dedicated workspaces.
![eu-device-requirements-class-III]() Device Class III Requirements
Device Class III Requirements
 Device Class III Requirements
Device Class III RequirementsClass III devices are considered high-risk and are subject to the most stringent requirements, including clinical evaluation of the device. This risk class includes devices like pacemakers, prosthetic heart valves, surgical mesh, breast implants, and other devices that require permanent monitoring throughout their lifetimes.
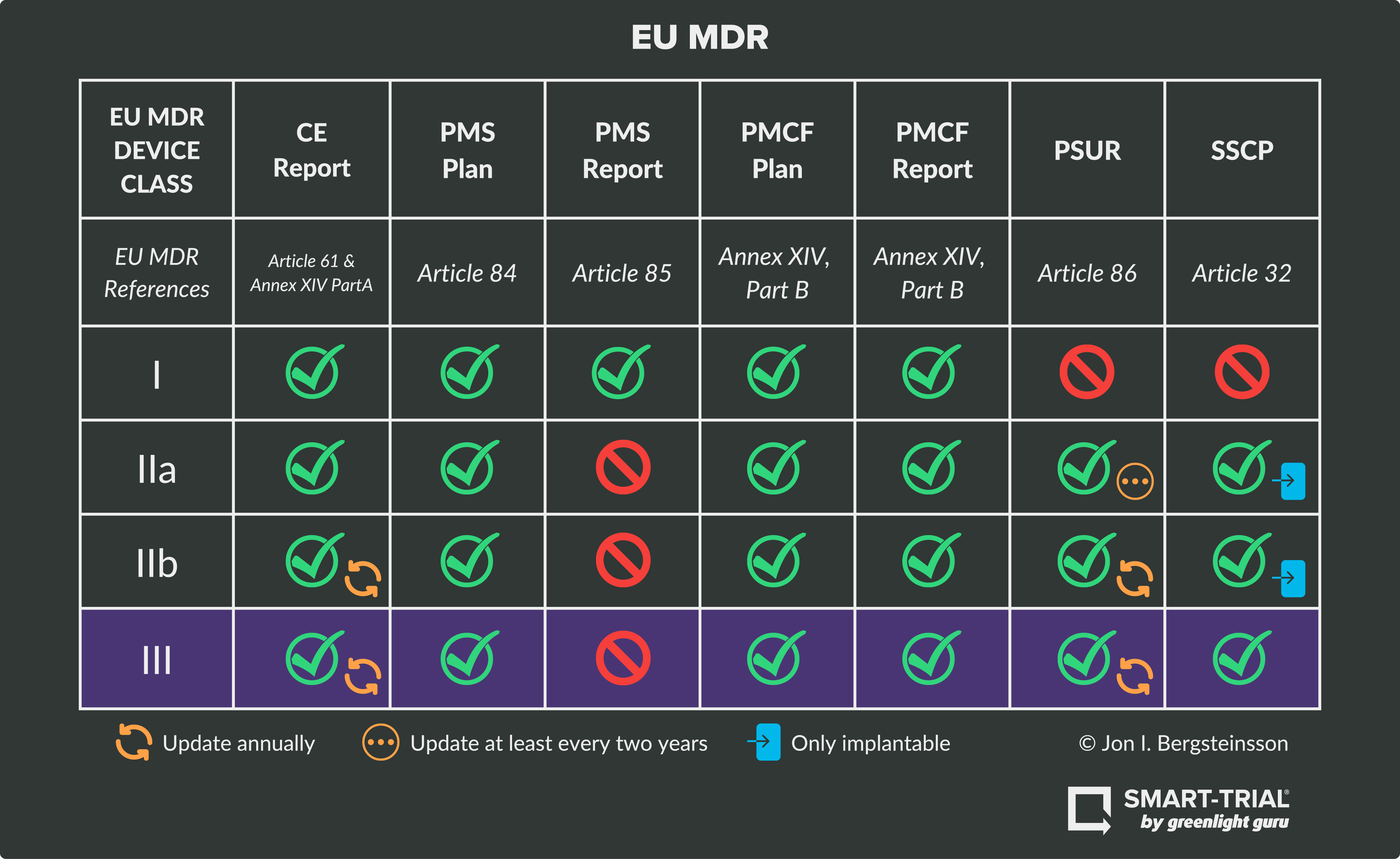
As seen in the graphic above, Class III devices must fulfill obligations and technical requirements for:
-
Clinical Evaluation Report (CER)
-
Post-market Surveillance Plan (PMS Plan)
-
Periodic Safety Update Report (PSUR)
-
Post-market Clinical Follow-up Plan (PMCF Plan)
-
Post-market Clinical Follow-up Report (PMCF Report)
-
Summary of Safety and Clinical Performance (SSCP)
NOTE: Class III devices DO NOT require a PMS report.
CE Report
What is it? The clinical evaluation report (CER) is documentation of the clinical evaluation that is required of every medical device sold in the EU. Its purpose is to prove your device performs as intended without compromising the safety of its end users.
How is it regulated under EU MDR? Article 61 of EU MDR requires every medical device manufacturer to document the clinical evaluation of their device in a CER.3 This requirement is expanded upon in Annex XIV Part A, which states:
The results of the clinical evaluation and the clinical evidence on which it is based shall be documented in a clinical evaluation report which shall support the assessment of the conformity of the device.
MDCG-2020-5 and MDCG 2020-13 are guidance documents for clinical evaluations, and contain instructions and specific requirements for the clinical evaluation report.5
How to comply? MDCG-2020-5 states that your CER should contain sufficient information for anyone reading it to understand the search criteria, the included data, and all assumptions made as well as all conclusions reached.
MDCG-2020-13 provides a sample table of contents, which indicate the CER should include:
-
Administrative particulars
-
Medical device name model and type
-
Manufacturer(s) name and SRN
-
Notified body
-
Type of assessment
-
Intended purpose
-
Check of clinical evaluation report authors
-
Reviewers involved in notified body assessment of the clinical evaluation
-
Device information, including:
-
Description
-
Classification
-
Clinical evaluation plan
-
Information materials supplied by the manufacturer
-
Common specifications and harmonised standards applied
-
Demonstration of equivalence
-
State of the art
-
Clinical literature review
-
Clinical investigations and related documentation
-
PMS, PMCF and the plan for updates
-
IFU, SSCP, labeling and other information supplied with the device
-
Summary of all available data and conclusions
-
Overall Conclusions
Lastly, the guidance states that the report’s contents shall be cross-referenced to supporting documents, and to their locations in your technical documentation.
When do I submit or update the CE Report? The CE report for Class III devices is intended to be a living document, and therefore must be updated annually, as well as at any other time it is relevant to do so, such as when significant changes occur.
How can Greenlight Guru help you manage the entire clinical evaluation process? A successful clinical evaluation report hinges on the quality of clinical data and the processes for collecting and analyzing it. To do so, researchers use Case Report Forms (CRFs) completed by investigators for each patient during the investigation.8
In the past, CRFs from a clinical study would be completed on paper, packed into boxes, and shipped from the study location to a secondary location for analysis. But today, a growing number of medical device companies are going paperless and using web-based Electronic Data Capture (EDC) systems to collect, store, and manage clinical data.9
Software-based EDC systems give clinical study managers the ability to streamline the data collection process, capture more accurate data, and enhance data security and accessibility - all while saving time and reducing the cost of completing a clinical study. EDC systems are also an essential pairing with a well-developed quality management system to ensure that best practices are being followed.
With Greenlight Guru Clinical, medical device companies can create customized electronic case report forms (eCRF) and digitize data collection for clinical investigations, in-human studies, and post-market surveillance activities.7
Post-market Surveillance Plan
What is it? The post-market surveillance (PMS) plan is part of a device’s required technical documentation, detailing your strategy for continuously monitoring and collecting data and safety information on the device. The plan is part of the requirements for a PMS system, and is intended to outline the criteria for the benefit-risk assessment of the device and processes for:
-
Collecting and analyzing data
-
Addressing submitted complaints
-
Communicating data to regulatory bodies and users
-
Implementing corrective actions on devices
Additionally, the PMS plan is used to determine whether or not a post-market clinical follow-up plan (PMCF) is required.
Where is it in the EU MDR? Article 84 requires that the PMS system is based on a plan.4 However, the details of the plan are specified in Annex III, 1.1.
How to comply? A compliant PMS plan should consider information concerning serious incidents, records of non-serious incidents, available data on side-effects, information from trend reports, any feedback or complaints provided by users, distributors or importers of the medical device, and publicly available information about similar devices.
Annex III of EU MDR requires the following topics to be covered in the PMS plan:
-
A systematic and proactive process for collecting information (user feedback, reports, etc.)
-
Appropriate statistical and analytical methods of assessing collected data
-
Threshold values for assessing benefit-risk and effective risk management
-
Appropriate methods and tools for investigating complaints and other feedback collected in the field
-
Methods for managing events subject to the trend report
-
Protocols for communicating effectively with the competent authorities, notified bodies, economic operators, and users
-
Reference for procedures to fulfill the manufacturer's obligations for PMS system, PMS plan, and PSUR
-
Procedures and systems for implementing corrective actions when required
-
Effective tools to trace and identify devices for which corrective actions may be needed (traceability of potentially defective products in case a recall is needed, for example)
When do I submit or update the PMS plan? The PMS plan must be established and documented prior to placing the medical device on the market for the first time, and updated as necessary during its lifecycle.
How can Greenlight Guru help you with your PMS plan? Post-market surveillance is a defining pillar of MDR. Throughout the regulation, we see many ways the PMS system touches or feeds into every part of the total product lifecycle, including the ongoing risk management and clinical evaluation processes.
If that wasn’t enough, the PMS plan must also address the processes used to collect and assess the massive amounts of data produced for a single device. This can be cause for concern among manufacturers, especially for those using a paper-based data collection process which carries high risk of missing and erroneous data.
Greenlight Guru Clinical makes it much more efficient to collect PMS data with direct data capture (DDC).6 Our electronic data capture (EDC) software lets medical device manufacturers seamlessly enter clinical data contemporaneously (during visits) or at a later point in time. Designed to fit MedTech needs, you don’t need technical skills to get started. Our ready-to-use templates, modules and features will help you to easily design the optimal clinical study.
Another benefit of our EDC software is that it allows clinicians to collect patient reported outcomes electronically on any device, thanks to bring-your-own-device (BYOD) capabilities. Our clients make full use of the ePRO capabilities by tailoring it to their clinical study needs as they solidify their device’s PMS plans.
Periodic Safety Update Report
What is it? The Periodic Safety Update Report is a summary of the results of postmarket surveillance activities as well as the conclusions that manufacturers have drawn from those results. If the manufacturer has taken any corrective or preventive actions (CAPAs), a description and rationale for the actions must also be included in this report.
The Periodic Safety Update Report is part of a device’s technical documentation, and it has to be updated throughout the device’s lifecycle. The PSUR is initially submitted to a notified body during the device’s conformity assessment audit, but from then on it must be updated either annually or biennially.
Where is it in the EU MDR? The full requirements for your PSUR can be found in Article 86 of MDR,11 or Article 81 of IVDR.12
How to comply? PSURs are identical to PMSRs with a few additions - manufacturers must publish the conclusion of the benefit-risk determination, main findings of the post-market clinical or performance follow-up, sales volume, and estimated user population characteristics and usage frequency.
When to submit or update? Manufacturers of Class III devices shall update the PSUR when necessary and at least every two years. That PSUR shall, except in the case of custom-made devices, be part of the technical documentation as specified in Annexes II and III.
How can Greenlight Guru support your PSUR? The Periodic Safety Update Report is not just a data dump or a chance to check some boxes. It is intended to be an analysis of the postmarket data your company has actively collected. It’s a meaningful, regularly updated report that should help both regulators and internal stakeholders understand the safety and efficacy of your device during its time on the market.
In order to help readers understand that this marketed device is safe, effective, and successful at fulfilling its intended purpose, you need to provide rationale in the form of data-driven and meaningful conclusions as a result of your postmarket surveillance and clinical followups. This can be difficult for companies — what if your argument doesn’t work? What if your conclusions are unconvincing, or customer data on usage are spotty?
This concern demonstrates the need for using validated questionnaires. Validated questionnaires are especially useful when you need to gather data for regulatory purposes because they are scientifically tested. This means that the results from those questionnaires can provide reliable information on the topic of interest - if applied correctly. In addition, it also simplifies the justification of your data collection method, to why you are asking specific questions.
With Greenlight Guru Clinical, creating custom questionnaires and forms to use in a PMCF survey or clinical investigational context often returns a more complex dataset. Standardized questionnaires are helpful, because they provide sponsors with a structured dataset that is easy to analyze. It minimizes the need to clean data or merge open-ended or text-based answers. In the end it can save a great amount of time in creating a competent and thorough PSUR.
Post-market Clinical Follow-up Plan
What is it? The Post-market clinical follow-up plan (PMCF) specifies the methods and procedures used to proactively collect and evaluate clinical data on a device’s performance and safety. As part of your technical documentation, PMCF is connected to and used to update the PMS plan and CER. It also serves as the template for your PMCF report.
The goal of PMCF is to identify previously unknown side-effects, assess emergent risks, and prevent off-label misuse. This data can be captured through general PMCF activities, such as gathering feedback from end users and information from scientific literature.
However, in order to substantiate the data, it often is necessary to use specific PMCF activities—higher-level operations which can be used to scientifically illustrate safety and clinical performance, often based on case-specific data.10
These PMCF activities include:
-
PMCF studies (or observational or non-interventional clinical investigations)
-
Post-market interventional clinical investigations
-
Evaluation of data from suitable registries
-
Investigator initiated studies
-
Case cohorts
-
Other subject-specific clinical data collection activities
Where is it in the EU MDR? Post-Market Clinical Follow-Up activities are required by Part B of MDR’s Annex XIV, and further guidance and a template are available in the guidance document MDCG 2020-7.13
How to comply? The PMCF is a formal activity and cannot be performed in an ad hoc manner. You’ll need to create a post-market clinical follow-up plan that outlines the process you intend to use to gather and evaluate data for your PMCF evaluation report.
Generally speaking, your PMCF plan will have seven sections:
-
The manufacturer’s contact details
-
A description and specification of the medical device being studied
-
The activities related to the PCMF (these are both the general and specific methods and procedures you’re using)
-
References to any relevant parts of the technical documentation
-
An evaluation of clinical data for equivalent or similar devices
-
References to any applicable common specifications, harmonized standards, or guidance documents
-
Estimated date of the PMCF evaluation report
When to submit or update the PMCF plan? By definition, Post-Market Clinical Follow-up activities occur after a medical device has been placed on the market. However, PMCF plans are part of the PMS system, and should there be any corrective actions taken as a result of the clinical studies, the PMCF should be updated to reflect those changes.
How can Greenlight Guru help you with your PMCF plan? A common pitfall for medical device companies planning their PMCF activities are communication breakdowns, such as the need for approval of ethical committees, as well as data silos that prevent key stakeholder opinions from staying connected. PMCF planning is a time-consuming task and the increased cost from this step is often a surprise for medical devices companies with limited PMCF experience.
With Greenlight Guru Clinical, you can use a personalized PMCF plan template that is reusable across products and markets. Your team can make generic product-related questions for safety and performance. Also, you can select specific scales to track both.
You can also benefit from the centralization of both your clinical data and your quality management system data, thanks to Greenlight Guru’s industry-leading eQMS solution. Linking all these items in a single workflow environment means you won’t be looking for missing documentation, lost signatures, or any of the other frustrations that come with outdated paper-based tracking.
Post-market Clinical Follow-up Report
What is it? The Post-market Clinical Follow-up Report is the structured summarization of the resulting data from the PMCF plan. Recording and reporting of adverse events and serious adverse events is a key aspect of a successful clinical investigation and staying on top of regulatory changes is a must.
Where is it in the EU MDR? PMCF reports are required under Annex XIV Part B of the MDR.2 Further guidance can be found in MDCG 2020-8, which outlines the reporting requirements and provides a template.14
How to comply? PMCF activities conducted for your device must follow the latest requirements for GCP (Good Clinical Practice).16 As part of GCP, you are required to notify competent authorities of serious incidents that were previously left undocumented, such as previously unknown side-effects. Additionally, you’ll need to clearly describe this incident in the product documentation prior to introducing your device into the EU market.
When to submit or update? Just like PMCF plans, the PMCF reports will naturally come during your post-market surveillance phase, and will subsequently require being updated whenever appropriate.
How can Greenlight Guru help you with your PMCF report? One of the critical quality requirements for both clinical investigations and PMCF under the MDR is compliance with GCP. To ensure that the data collected in a PMCF survey complies with the quality standards of the EU MDR, sponsors have to use solutions that can fulfill the ISO 14155:2020 requirements for electronic data capture.17
This means that if you use survey software that does not comply with ISO 14155:2020, you risk not being able to use the PMCF survey data in your clinical evaluation report. Which then can potentially lead to a loss of CE mark.
To avoid this, you’ll need to ensure your EDC software provider can:
-
Provide documentation of verification and validation according to the requirements specified in ISO 14155:2020
-
Document data traceability from source to sponsor
-
Comply with all standard GCP principles when collecting, viewing, editing, and exporting data
-
Store the data according to industry standards on general security and permission control
-
Comply with GDPR requirements for informed consents and data protection
With Greenlight Guru Clinical, you can rest assured knowing that our EDC solution is pre-validated and compliant with both ISO 14155:2020 and GCP. It’s also why our medical device-specific solution is designed to stay up-to-date with the latest FDA, ISO, and EU MDR guidelines for post-market and clinical study needs.
Summary of Safety and Clinical Performance
What is it? An SSCP/SSP is an external document that incorporates information related to a medical device, such as general information, a summary of the clinical data collected from the device, or possible therapeutic alternatives. SSCP is intended for devices which are higher risk or invasive, specifically:
-
Implantable Class IIa devices
-
Implantable Class IIb devices
-
All Class III devices
The main purpose of the SSCP/SSP is to provide both healthcare workers and patients access to current clinical, safety, performance and other types of data from the medical device; as this aligns with the MDR/IVDR philosophy of providing a more transparent and robust regulatory framework.
Where is it in the EU MDR? The requirement and general definition is found in MDR, under Article 32. For further instruction, consult the guidance doc MDCG 2019-9 – Rev.1.15
How to comply? The SSCP/SSP must be objective, including both favorable and unfavorable data. It should be based on the device's technical documentation and constantly revised to prevent outdated information.
MDCG 2019-9 – Rev.1 states the SSCP must at minimum contain sections on:
-
Device and manufacturer general details – including UDI details
-
Intended purpose
-
Indications, contraindications and target population
-
Description of the device, its components, medicinal products and any previous version of the device
-
Residual risk, warning and precautions details
-
Summary of the Clinical Evaluation of the device, emphasizing the PMCF evaluation and pointing out conclusions related to safety and performance
-
Description of necessary training and qualifications
-
Reference to harmonized standards and common specifications applied
-
Revision history
When to submit or update? Implantable Class III devices must prepare SSCPs annually, and may include updates from the Post-Market Clinical Follow-up (PMCF) and the Product Safety Update Report (PSUR) as part of its ongoing lifecycle.
How can Greenlight Guru support your SSCP? The SSCP needs to translate technical documentation into messaging that anyone can understand, not just those trained to be medical device experts.
This document will be a guide for providers and patients alike, and therefore can’t be viewed as a piece of marketing material. The information must be objective, sometimes even negative, while still balancing transparency with the needs for user privacy. In short—writing these documents is an art.
The solution for this problem is to simplify the process with templates that work well, together. With Greenlight Guru Clinical, we deliver out-of-the-box compliance with ISO 14155:2020, along with user-friendly eCRF design templates, GDPR-compliant data export, and support for many different study designs and protocols.
And because the SSCP plays so heavily into ISO requirements for continuous improvement and quality management systems, the Greenlight Guru EDC software solutions pair beautifully with our eQMS solution and workspace.

Why you need a best-in-class EDC and QMS partner
Regardless of your device class, all MedTech companies need quality clinical processes that produce and collect high-quality clinical data. And for good reason—clinical processes are continuous and critical to ensure your device is safe and effective throughout the product lifecycle. It’s also the reason why you need the right partner for your EDC and QMS solution.
With Greenlight Guru, Clinical you’ll leverage the only Electronic Data Capture (EDC) platform designed for MedTech manufacturers that fits the future of the MedTech industry and its regulatory framework. Users also gain access to an in-depth, pre-validated toolbox for Post-Market Clinical Follow-Up (PMCF), Post-Market Performance Follow-Up (PMPF), Clinical Investigations, and Clinical Performance Studies.
And of course, Greenlight Guru offers the only Quality Management Software designed by medical device professionals specifically for medical device professionals. Our comprehensive, out-of-the-box solution is based on the latest industry standards and regulations, as well as best practices of medical device market leaders.
Want to learn more about Greenlight Guru Clinical? See a personalized demo here →
References
-
Regulation (EU) 2017/745. 2017. Annex VIII. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0745.
-
Regulation (EU) 2017/745. 2017. Annex XIV Part B. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0745.
-
Regulation (EU) 2017/745. 2017. Article 61. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0745.
-
REGULATION (EU) 2017/745. 2017. Article 84. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0745.
-
MDCG 2020-5 Clinical Evaluation - Equivalence: A guide for manufacturers and notified bodies. 2020.
https://health.ec.europa.eu/system/files/2020-09/md_mdcg_2020_5_guidance_clinical_evaluation_equivalence_en_0.pdfMDCG 2020-13 Clinical Evaluation Assessment Report Template. 2020.
https://health.ec.europa.eu/system/files/2020-07/md_2020-13-cea-report-template_en_0.pdf -
“Medical Device Clinical Software | Greenlight Guru Clinical.” https://www.greenlight.guru/clinical-electronic-data-capture-software.
-
Jóhannesson, Páll. The Essential Guide to Electronic Case Report Form (ECRF) for Medical Devices. August 30, 2022. https://www.greenlight.guru/blog/electronic-case-report-form.
-
Latha, MS, Shantala Bellary, and Binny Krishnankutty. 2014. “Basics of Case Report Form Designing in Clinical Research.” Perspectives in Clinical Research 5 (4): 159. https://doi.org/10.4103/2229-3485.140555.
-
Krishnankutty, Binny, BR Naveen Kumar, LathaS Moodahadu, and Shantala Bellary. 2012. “Data Management in Clinical Research: An Overview.” Indian Journal of Pharmacology 44 (2): 168. https://doi.org/10.4103/0253-7613.93842.
-
Bergsteinsson, Jon Ingi. Selecting the Ideal PMCF Activity (blog). July 17, 2022. https://www.greenlight.guru/blog/selecting-the-ideal-pmcf-activity.
-
Regulation (EU) 2017/745. 2017. Article 86. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0745.
-
Regulation (EU) 2017/746. 2017. Article 81. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0746
-
MDCG 2020-7 Post-Market Clinical Follow-up (PMCF) Plan Template. A Guide for Manufacturers and Notified Bodies. 2020. https://ec.europa.eu/docsroom/documents/40905.
-
MDCG 2020-8 Post-Market Clinical Follow-up (PMCF) Evaluation Report Template. A Guide for Manufacturers and Notified Bodies. 2020. https://ec.europa.eu/docsroom/documents/40906.
-
MDCG 2019-9 Rev.1 Summary of Safety and Clinical Performance a Guide for Manufacturers and Notified Bodies. 2022. https://ec.europa.eu/health/system/files/2022-03/md_mdcg_2019_9_sscp_en.pdf.
-
ICH. 2016. Guideline for Good Clinical Practice E6(R2). https://www.ema.europa.eu/documents/scientific-guideline/ich-e-6-r2-guideline-good-clinical-practice-step-5_en.pdf
- Rish, Tom. 2021. How to Plan and Conduct an ISO 14155-Compliant Clinical Investigation (blog). May 31, 2021. https://www.greenlight.guru/blog/iso-14155.
About the Authors

Laura Court, Medical Device Guru, Greenlight Guru
Laura Court is a Medical Device Guru and Mechanical Engineer who loves learning about new technology and sharing her experiences with others. She has experience in New Product Development and Manufacturing with both small and large companies. Having managed products through their full product lifecycle into post market support, Laura understands the amount of work required to build out all the documentation required for regulatory approval and support. Laura's knowledge, experience and passion with medical devices is evenly matched with how much she enjoys helping customers work efficiently through the design and development process to bring safe, high quality products to market.
.png)
Jón Bergsteinsson, co-founder, Greenlight Guru Clinical
Jón Ingi Bergsteinsson, M.Sc. in Biomedical Engineering, is the co-founder of Greenlight Guru Clinical, where he helps MedTech companies bridge the gap between their devices and clinical data. As the technical founder of Greenlight Guru Clinical he previously served as the CTO where he paved the way for the platform’s quality standards, data security, and compliance.
FREE eBOOK:
Ultimate Guide to Device Class Requirements under EU MDR
Ultimate Guide to Device Class Requirements under EU MDR






