.png?width=800&height=400&name=Medical%20Device%20Clinical%20Trials%20Regulatory%20Pathways%20%26%20Study%20Types%20Explained%20(1).png)
What is a clinical trial?
In both the US and the EU, medical devices may be required to undergo a clinical trial before they can be placed on the market. A clinical trial is a systematic assessment of the device's safety and/or efficacy that uses human participants, and it's a requirement requirement for certain risk classes. For some products, including in vitro diagnostics, device trials may also be used to evaluate device performance, clinical use, adverse events, and the evidence needed for regulatory review.
-
In the EU, all Class III and Class IIb implantable devices must undergo clinical investigations according to EU MDR.
-
In the US, all Class III devices are required by the Food and Drug Administration (FDA) to undergo clinical investigations as part of premarket approval (PMA).
With that in mind, let's take a look at the different stages and designs of medical device clinical trials and the regulations surrounding them.
NOTE: You may see clinical trials referred to as "clinical studies" or, more commonly in the medical device industry, "clinical investigations." A medical device clinical investigation may also be described as a device trial, depending on the regulatory setting and audience. These terms are all synonymous and can be used interchangeably.
How are medical device clinical trials initiated?
Because clinical trials involve human participants, medical device companies must perform preclinical testing and research on their product before even applying for a clinical trial.
Preclinical activities determine whether a device is safe and effective enough for use with human subjects, and include steps like:
-
Bench testing
-
Technical testing
-
Computer simulations
-
Animal studies
Once the manufacturer believes their device is ready for clinical trials, they must first get approval for their proposed investigation. At this stage, teams typically define the clinical trial protocol, expected sample size, control group strategy, device performance endpoints, adverse events reporting process, and Data Analysis plan. The processes for getting approval and initiating a clinical trial in the EU and US are different, so let's take a look at each.
Clinical trial regulatory pathways in the US
In the US, medical device manufacturers that want to pursue a clinical trial must obtain an Investigational Device Exemption (IDE). Under the Federal Food, Drug, and Cosmetic Act and related FDA Guidance, an IDE allows an investigational device to be used in a clinical study to collect safety and effectiveness data. Only once the IDE has been approved can a device that has not yet received market approval be tested on human subjects.
There are exceptions to the IDE submission, which include certain low-risk diagnostic devices, some in vitro diagnostics, certain Class I medical devices, some Class II Medical Devices, as well as devices that are determined to be non-significant risk (NSR).
If a device is granted an IDE, the clinical investigation must still be reviewed by an Institutional Review Board (IRB). Clinical trials are generally performed within an institution, such as a hospital, and an IRB is an additional layer of scrutiny that the institution provides to ensure the study meets its standards. The study may begin only once the IRB has approved it and FDA has approved the IDE application.
Clinical trial regulatory pathways in the EU
In the European Union, EU MDR has 20 articles outlining the requirements for clinical investigations of medical devices, spanning articles 62 through 82. These requirements help regulatory bodies evaluate whether a medical device clinical investigation has been designed, authorized, conducted, recorded, and reported appropriately. Within these articles, the regulation lays out three regulatory pathways manufacturers can take:
-
Article 62 covers investigations that are performed in order to demonstrate conformity and obtain a CE marking. This is the pathway medical device companies will use if their device classification (for Class III or Class IIb implantables) requires a clinical investigation.
-
Article 74(1) covers the regulatory pathway for devices that already have a CE marking if the parameters of the investigation are within the device's intended purpose. In other words, if you are conducting a clinical investigation as part of your Post-Market Clinical Follow-Up (PMCF), then you will be guided by Article 74(1).
-
Article 82 covers clinical investigations that are not being performed in order to demonstrate conformity. Additionally, the Member State in which you hold your study may have relevant national provisions for you to follow.
For in vitro diagnostics, manufacturers should also confirm whether IVDR performance study requirements apply. While MDR and IVDR differ, both frameworks require teams to understand the relevant regulatory setting before initiating clinical research or submitting evidence for regulatory review.
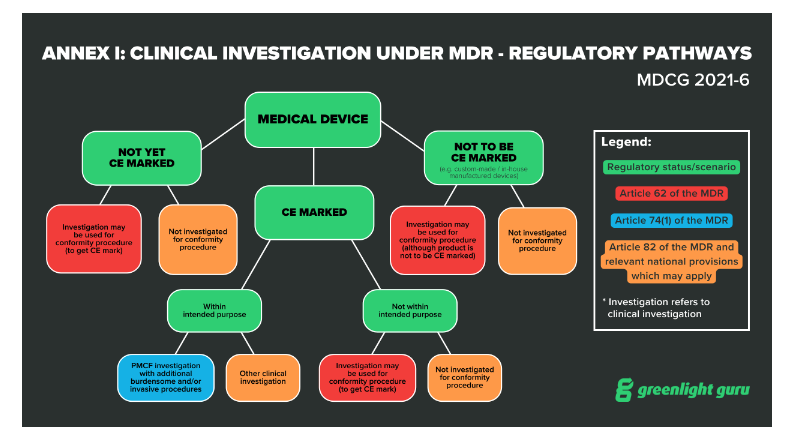 Annex I: Clinical Investigation under MDR - regulatory pathways, MDCG 2021-6 (Source)
Annex I: Clinical Investigation under MDR - regulatory pathways, MDCG 2021-6 (Source)
Before initiating a clinical trial in the EU, you'll also need a CIV-ID and approval from the relevant competent authority. The CIV-ID is an EU specific tracking number that competent authorities in any Member State can use to identify and track your clinical investigation.
.png)
What are the different stages and types of medical device clinical trials?
Clinical trials may be carried out during both the premarket and postmarket phases of the device lifecycle. These device trials can support clinical development, regulatory submissions, post-market surveillance, and long-term evidence generation for medical devices. The graphic below includes the many different types of clinical activities, including clinical trials, medical device manufacturers may carry out during the pre-market and post-market phases.
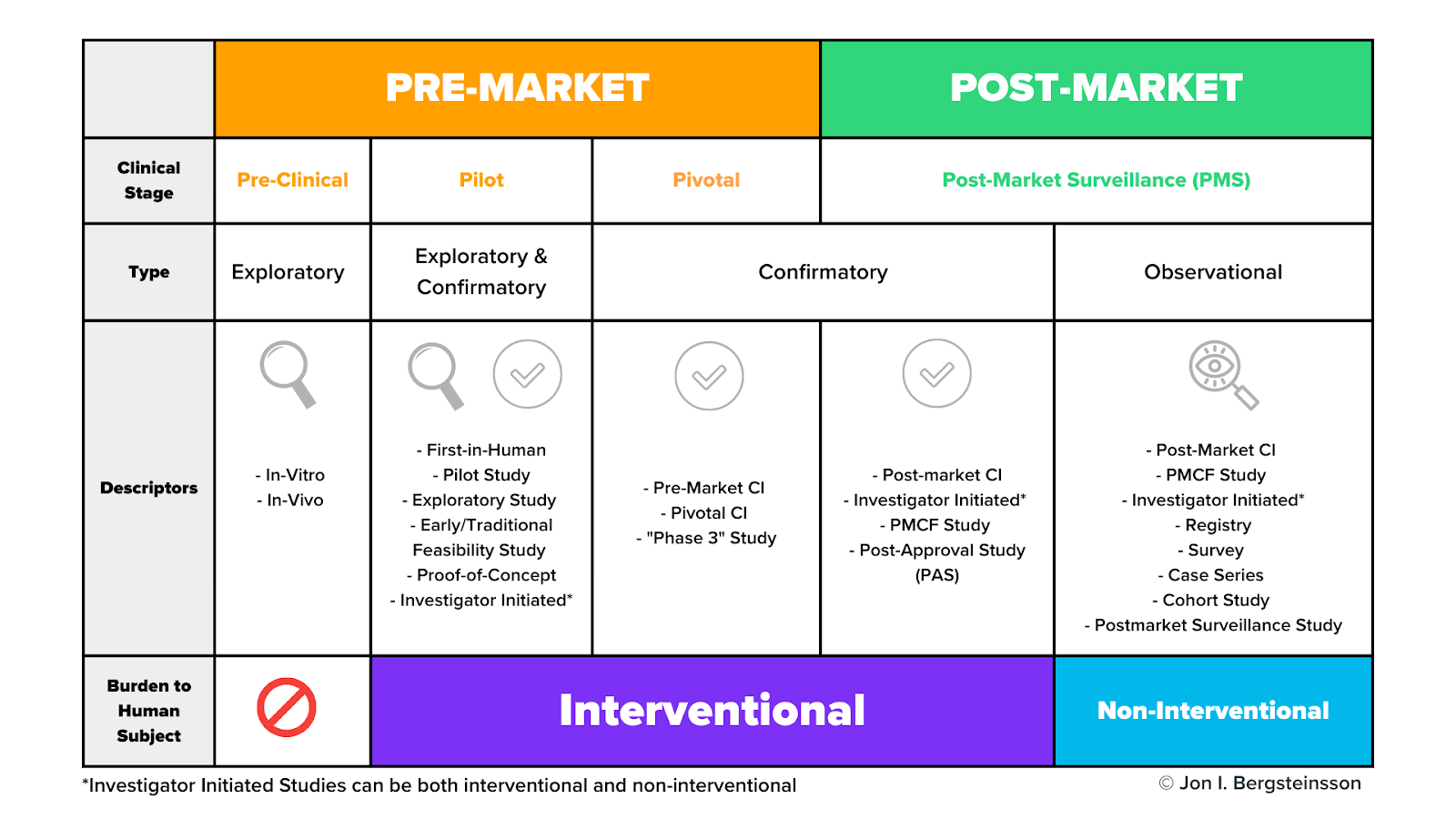
Clinical trials may occur during the pilot stage, the pivotal stage, or during post-market surveillance. As I mentioned earlier, pre-clinical activities do not use human subjects.
As we dig in here, don't get too hung up on the study descriptors in this graphic. Many of these terms are interchangeable, and different descriptors are often used in different markets to describe the same thing. For now, let's focus on the general types and stages of studies and their burden to human subjects.
What are pilot studies?
Pilot studies occur early in device development, often before the device design has been finalized. Pilot studies are used when nonclinical testing is unable to provide preliminary information on device functionality and clinical safety. These will be conducted with a very small number of patients, often 10 or fewer. For some device trials, pilot data may also help refine sample size assumptions, determine whether a control group is feasible, and clarify how adverse events or an adverse event rate will be captured in a later pivotal study.
The purpose of pilot studies is to gain a broad range of information that may be used to:
-
Identify modifications to the device or procedure
-
Optimize operator technique
-
Refine the intended use population
-
Refine nonclinical test plans or methodologies
-
Develop subsequent clinical trial protocol and clinical study protocols
The data you gain from a pilot study may then be used to help you design a pivotal study later on.
What are pivotal studies?
A pivotal study is used to gather definitive evidence of the safety and effectiveness of your medical device for a specific intended use. These studies generally use a larger number of subjects than pilot studies, and you'll use the results of your pivotal study to gain regulatory approval for your device. In many device trials, the pivotal study design will define the sample size, control group, device effectiveness endpoints, device performance measures, adverse events reporting requirements, and statistical approach before enrollment begins.
Keep in mind, a pivotal study does not necessarily need to be preceded by a pilot study. The types of clinical activities you carry out will depend on your device and the regulatory pathway you're taking.
Do clinical trials happen during post-market surveillance?
As you can see from the graphic, the post-market surveillance stage includes both confirmatory and observational types of clinical activities.
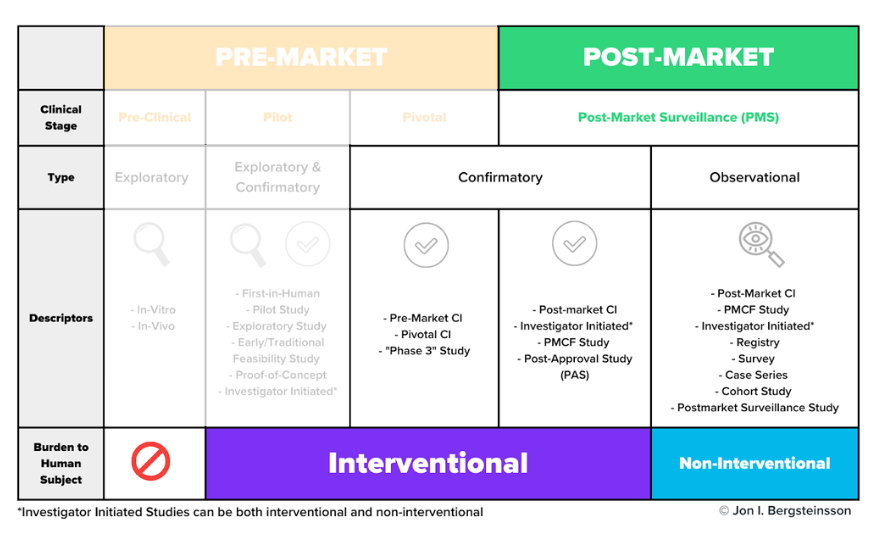
While it may seem odd that you would need to perform a confirmatory study after receiving approval to place your device on the market, this is not an irregular occurrence. For example, EU MDR includes a distinct regulatory pathway, Article 74(1), for conducting a clinical investigation as part of your PMCF.
These post-market surveillance studies may be conducted for a number of reasons, including to confirm the safety and efficacy of the device once it's on the market or to answer questions about the long-term safety or performance of the device. They may also help manufacturers monitor adverse events, medical device reports, real-world device performance, and safety signals that emerge during routine clinical use.
How are observational clinical activities conducted?
Many post-market clinical activities are categorized as "observational" and they use non-interventional methods to collect data.
-
In interventional studies, such as a pivotal study, someone is actively recruiting participants. For example, a physician may ask a patient who may benefit from a certain device if they would like to volunteer for that study. In other words, they are intervening in the normal clinical pathway the patient would follow.
-
In non-interventional studies, there is no intervention in the clinical pathway, merely observation. For example, a physician prescribes a treatment they believe the patient needs (the normal clinical pathway),and then asks the patient if they would agree to share the data related to their treatment as part of an observational study. Even when there is no randomized control group, observational clinical research should still define the sample size, data collection process, adverse events monitoring approach, and Data Analysis plan.
Remember, some devices may need clinical data from all of these categories, but many will not. For example, low risk devices relying on well-known technology may not require any clinical investigations on your part.
Greenlight Guru Clinical helps you streamline clinical data collection for your medical device
This may seem like a complicated topic, but if you break it down by the stages of the device lifecycle and the type of clinical activity, you have a roadmap for how you'll obtain the necessary clinical data for your device.
And when it comes time to begin collecting that data, you'll need a flexible, modern platform that can streamline data collection from any and all of your clinical activities.
Greenlight Guru Clinical is that platform. Whether you're gathering data in clinical studies, performance studies, PMCF/PMPF studies, surveys, registries, cohorts, case series, or device trials for medical devices, our Electronic Data Capture solution allows you to collect and manage it all with ease. From clinical trial protocol setup to adverse events tracking, sample size planning, data analysis, and post-market clinical research, Greenlight Guru Clinical helps teams manage the evidence needed for regulatory review. Even better, it comes fully validated out of the box per ISO 14155:2020.
When you are ready to evaluate EDC vendors for your investigation, our EDC selection guide covers the 7 criteria, vendor question bank, and how to build the internal business case.
Ready to learn more? Contact us today for a customized demo.
BONUS RESOURCE:
15-in-1 Clinical Investigations Content Bundle
15-in-1 Clinical Investigations Content Bundle

.png?width=790&name=15-in-1%20clinical%20investigations%20content%20bundle%20(new).png)



