
Would things be simpler in the medical device regulatory space if there was global harmonization across all markets?
For at least the last two to three years, there has been buzz around the medical device industry about a possible harmonization between the FDA Quality System Regulation (QSR) and ISO 13485, the international standard for quality management systems of medical devices, with indications showing that it’s an active initiative of FDA.
In many ways, this could be a positive step for medical device companies, especially manufacturers who already have ISO 13485 certification and plan to enter the US marketplace. In other ways, it may not make much of a difference for manufacturers who already comply with FDA 21 CFR Part 820, since the two sets of requirements follow each other very closely.
Is global harmonization on the horizon? What will it mean for you if it is? Let’s take a closer look:
FDA plans to harmonize QSR with ISO 13485
FDA first revealed plans to harmonize its QSR with ISO 13485 in 2018. Since then, there has been tremendous upheaval as a result of the coronavirus pandemic, resulting in the plan being delayed in order to address the public health crisis. the situation.
However, FDA has indicated that QSR and ISO 13485 harmonization remains a top priority on their agenda. Given this sentiment, which has been expressed by more than one of their top brass officials, we expect this initiative to be pushed forward sooner rather than later.
When the FDA first spoke of this idea to harmonize the regulations, they listed a few benefits. For example;
-
Opportunities to work more closely with foreign regulatory authorities and to facilitate regulatory convergence.
-
A globally harmonized QMS for medical device manufacturers.
-
In many areas, the FDA expressed that their QMS principles would be more robust in harmonizing with ISO 13485.
-
Stronger ties to risk management principles as found in ISO 14971.
There has been considerable work done on mapping the QSR and ISO 13485. An official technical report (AAMI TIR 102:2019) was published comparing the requirements of the two, showing the similarities and demonstrating the differences. It states:
|
This document was produced by AAMI QM/WG 01 comprised of representatives from the medical device sector. For the proper use of this TIR, readers should have full understanding of both 21 CFR 820 and ISO 13485:2016. Although the information contained in this document has been carefully considered, it is up to the individual organization to ensure compliance with all regulatory requirements. |
As an example, see the table below, taken from the AAMI report, highlighting the sections in QSR and ISO 13485 on quality systems:
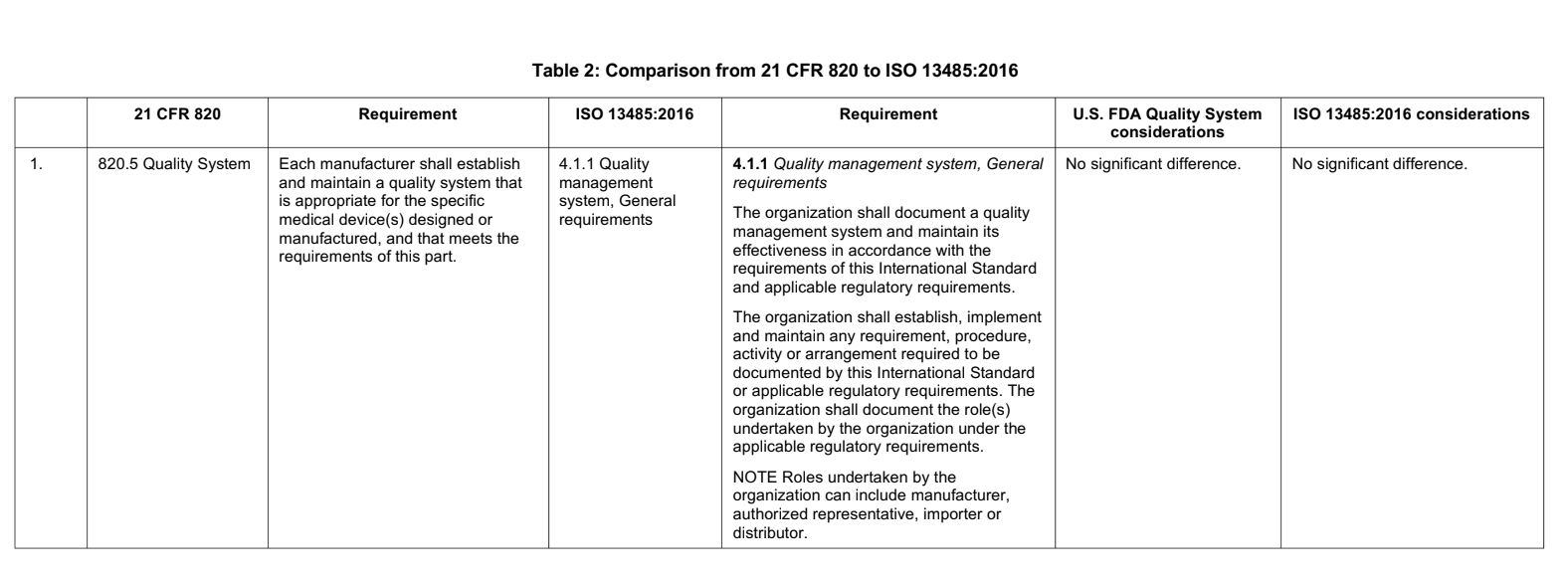
What are the differences between FDA QSR and ISO 13485?
If you take a high-level view of both FDA QSR and ISO 13485, they’re not that different. Both contain essentially the same requirements with respect to core expectations around medical device quality systems, processes, documentation and good manufacturing practices.
For example, both quality system requirements expect manufacturers to establish quality system procedures for:
-
Training
-
Purchasing
-
Device Master Record
-
Production & Process Controls
-
Labeling & Packaging
-
Receiving, Incoming, In-Process, Final Inspection
-
Identification & Traceability / Device History Record
-
Change Management
-
Nonconforming Material
-
CAPA
-
Management Responsibility
The international standard in many respects is a bit more prescriptive than QSR, but generally speaking, if your quality system meets the guidelines of the standard, then you should have no problem meeting QSR requirements.
At the moment, ISO 13485 certification is required for selling a medical device in certain markets, with the exception of the US. In the US, medical device manufacturers must meet the requirements of the QSR, but aren’t obligated to follow ISO 13485. On the other hand, if a medical device company wants to enter the Canadian market then ISO 13485 certification is required.
One of the key concepts introduced in ISO 13485:2016 was the application of a risk-based approach in establishing and maintaining a QMS. It references ISO 14971 with respect to managing medical device product risk.
The QSR doesn’t explicitly define risk-based requirements, however, the interpretation of the risk-based approach under ISO 13485:2016 is consistent with what the FDA expects under QSR.
The 2016 update of ISO 13485 actually brought a number of requirements in line with what the FDA was already mandating under its QSR. For example, document control and records management, particularly as both relate to protecting confidential information.
One of the main differences between FDA QSR and ISO 13485 is in how manufacturers are inspected or audited, respectively. While the FDA enlists their own inspectors, ISO audits are conducted by third-party registrars. Both have the same job though: checking and certifying that a medical device organization is following the applicable quality system requirements.
Importance of globally harmonized standard for medical device quality systems
Why are globally harmonized standards for medical device quality systems important?
For one thing, the hope is that harmonization can help to streamline and reduce some of the regulatory burden placed on medical device manufacturers that are selling into multiple markets around the world.
Rather than compartmentalizing documents and records according to which standard of regulation it goes with, you should be able to point to one set of records that satisfies all requirements.
The change will also help to further modernize regulations. While ISO 13485 has seen regular updates over the last couple of decades, the QSR has remained essentially unchanged since 1996. Consider all the technological changes that have taken place over that period of time - it’s imperative for the medical device industry to keep up!
Global harmonization has also been an active initiative of other regions. The roll-out of MDSAP (Medical Device Single Audit Program) has gained momentum in an effort to minimize audit and inspection burden.
For companies that are already ISO 13485:2016 certified, harmonization makes it simpler for them to enter the US market. The same applies to US manufacturers planning to enter foreign markets.
How to simplify the transition from 21 CFR Part 820 to ISO 13485
It is expected that with the transition to a harmonized standard for medical device quality systems, the impact on US medical device manufacturers will be minimal, although not without some challenges.
If your quality management system already complies with 21 CFR Part 820, then it should also align with ISO 13485, but of course not all medical device manufacturers are operating with a robust QMS!
How do you simplify the transition? With a QMS that is purpose-built for the medical device industry and is compliant with 21 CFR Part 820, ISO 13485:2016 and ISO 14971:2019.
Greenlight Guru’s medical device QMS software is built by medical device professionals, for medical device professionals and incorporates all of the latest regulatory best practices in the industry.
Meeting regulatory requirements has never been easier. Learn more about how Greenlight Guru can remove the regulatory burden so you can focus on True Quality medical devices by getting your free demo now →
Looking for a design control solution to help you bring safer medical devices to market faster with less risk? Click here to take a quick tour of Greenlight Guru's Medical Device QMS software



