
No matter where you plan on marketing your medical device, you’ll be required to perform a biological evaluation on your device before you receive regulatory approval.
However, not all medical devices will require the same sort of evaluation and testing, and if you’re trying to figure out what biological endpoints you need to evaluate, you should start with ISO 10993-1:2018 - Evaluation and testing within a risk management process.
Today, I want to walk you through the biological evaluation process—and show you the role that risk management plays in the decision of whether or not to perform specific tests on your device.
Create a biological evaluation plan (BEP)
The biological evaluation of a medical device is a complex process with plenty of nuance, which is why you need to start with a Biological Evaluation Plan (BEP).
Your BEP sets the stage for your evaluation by documenting everything you know about your device and serving as your initial risk assessment. Your BEP will highlight any knowledge gaps and residual risks that will require more information, and it will include your knowledge of:
-
The medical device. You’ll need to document everything about your device, such as its intended use, intended patient population, its materials and their processing, and the manufacturing process.
-
The recommendations from guidance. Using ISO 10993-1, you’ll be able to determine your device’s categorization, as well as the recommended biological endpoints for evaluation.
-
Any other available information. You’ll also need to document all the information currently available about this device, including any previous testing or clinical use history if the device is being resubmitted.
Categorize medical device based on type of contact and duration of contact
In order to determine which biological endpoints you need to evaluate, you first have to categorize your device according to the nature and duration of the device’s contact with the patient’s body.
Table A.1 in ISO 10993-1 contains a helpful visual matrix for the categorization process.
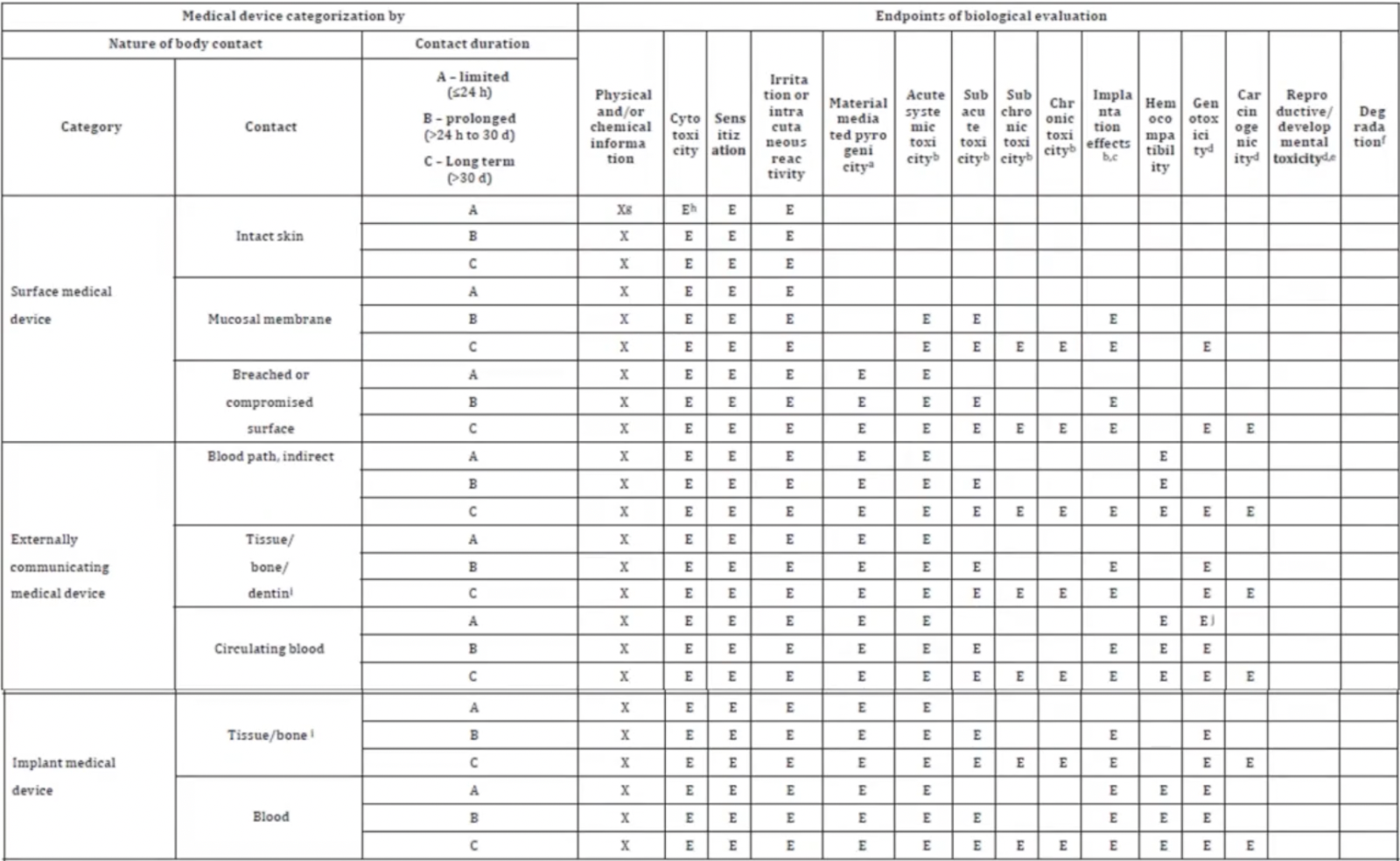 Table A.1 — Endpoints to be addressed in a biological risk assessment
Table A.1 — Endpoints to be addressed in a biological risk assessment
There are three initial categories your device may fall under:
-
Surface medical device
-
Externally communicating medical device
-
Implantable medical device
After choosing your initial category, you’ll then choose the type of contact your device has with the body (e.g., intact skin, mucosal membrane, breached or compromised surface) and the duration of the contact.
Contact duration is broken into three categories:
-
Limited (≤24 hours)
-
Prolonged (>24 hours to 30 days)
-
Long term (>30 days)
After choosing your contact duration, you’ll be able to look across the table and see the biological endpoints that should be evaluated for your particular device. Generally speaking, you’ll notice that the more invasive the contact and/or the longer the duration of the contact, the more endpoints need to be evaluated.
Use a risk-based approach to identify which endpoints need to be tested
Table A.1 includes all the biological endpoints that should be evaluated for each category. However, evaluated does not necessarily mean tested. ISO 10993-1 states that you might “reach the conclusion that no testing is needed if the material has a demonstrable safe history of use in a specified role and physical form that is equivalent to that of the medical device under design.”
You’ll notice that each cell in the column labeled “Physical and/or chemical information” is marked with an ‘X’. That’s because no matter the categorization of your device, it’s essential you know exactly what your product is made of, as well as the manufacturing process involved in making those materials and final device.
You’ll use that knowledge, in combination with the nature and duration of contact, to decide whether you need additional information (testing) to evaluate biocompatibility.
How to assess which endpoints you need to test
There are three important questions about your medical device that will help you get to the bottom of whether you need to test a specific biological endpoint:
-
What is it made of?
-
How is it made?
-
What is the available knowledge?
To give you an example, let’s say your device is a scalpel made out of one material: stainless steel. Using Table A.1, you identify it as an externally communicating device having limited contact with human tissue. Looking across the table, you’ll notice that there are five endpoints that must be evaluated, including “irritation or intracutaneous reactivity” and “sensitization.”
The question is, do you need to perform testing on all the endpoints marked for evaluation?
Now, stainless steel is a very well-known material. It’s been used for decades in medical devices and found to be nonreactive, which is why it’s often chosen for medical devices. In other words, there is a large amount of available information on stainless steel that you can draw upon.
However, you don’t want to forget the second question: How is it made?
The manufacturing process includes everything from receiving the raw material to the finished device. All of these steps could potentially leave some sort of residue on the final device that holds the potential for irritation or sensitivity.
For stainless steel, though, there is a well-known manufacturing process that progresses through the stages of machining, passivation, and sterilization. In particular, the passivation (making it even less reactive) process for stainless steel is highly standardized.
Given all of this, the residual risks are likely very low, and you could argue that testing for the endpoints of sensitization, irritation or intracutaneous reactivity, material mediated pyrogenicity, and acute systemic toxicity, are not necessary.
Keep in mind, if this were your medical device, you would still need a strong written justification of your reasoning for not performing these tests. The way you present your data and your justification is critical. You must be sure that anyone can read it and understand your rationale.
Greenlight Guru helps you integrate risk management into all your product development activities
Guidance like ISO 10993-1 makes it clear that risk management is more than just a checkbox activity—it should be woven into the very fabric of your medical device design, development, and manufacturing.
That’s why Greenlight Guru’s MedTech Suite is such an important tool for medical device companies. Our Risk Management Software reduces the stress of audits and inspections by integrating risk-based thinking into your entire quality ecosystem, allowing you to easily link and connect design controls and documents as risk control measures to mitigate and reduce risks throughout the entire product lifecycle.
If you want to see how our MedTech Suite can change your relationship with risk management, then get your free demo of Greenlight Guru today!
Looking for a design control solution to help you bring safer medical devices to market faster with less risk? Click here to take a quick tour of Greenlight Guru's Medical Device QMS software
FREE RESOURCE:
Biocompatibility Selection Criteria Chart
Biocompatibility Selection Criteria Chart






