
Medical device biocompatibility can be confusing, challenging, even frustrating for device professionals.
However, the biological evaluation of your device is one of the key components of a risk-based approach to medical device design and development.
There are many different types of biocompatibility tests, and the ones you perform will depend on the specifications of your device. So rather than explain each possible test, in this article I’ll provide an introduction to biocompatibility testing and review some of the most commonly asked questions on the subject.
FREE DOWNLOAD: Click here to get a printable PDF of the biocompatibility evaluation flow chart.
What is medical device biocompatibility and does it apply to my device?
Biocompatibility testing is used to ensure that a medical device which comes into direct or indirect contact with the human body does not produce an unacceptable adverse biological response.
For example, medical devices shouldn’t, either directly or through the release of their chemical components, cause adverse local or systemic effects. A local effect could include skin irritation or burns, while a systemic effect could include illness such as cancer, reproductive system effects or developmental effects.
Now, given that medical devices are made from an enormous variety of materials and used in many different situations, there are also many types of biocompatibility tests that a device may have to undergo. There are more than 20 different standards in the ISO 10993 family of biocompatibility testing standards, which cover topics such as:
- Cytotoxicity
- Sensitization
- Implantation
- Genotoxicity
- Carcinogenicity, reproductive and developmental toxicity
- Degradation assessments
However, ISO 10993-1, Evaluation and testing within a risk management process, is the most relevant standard for our overview of biocompatibility testing. ISO 10993-1 provides a general overview of biocompatibility testing and “is intended to describe the biological evaluation of medical devices within a risk management process, as part of the overall evaluation and development of each medical device”
FDA also has a guidance document, Use of International Standard ISO 10993-1, that provides more context for how they expect the standard to be applied by MedTech companies. Both the standard and FDA’s guidance emphasize that biocompatibility testing should be performed as part of a comprehensive risk management process, rather than as an afterthought.
Do you need to do biocompatibility testing?
The short answer is that biocompatibility testing is almost always required for medical devices that have contact with human tissue. Contact can be either direct or indirect.
- Direct contact means the device or device component comes into physical contact with body tissue.
- Indirect contact means a fluid or gas passes through the device or a device component prior to that fluid or gas coming into contact with body tissue.
The ISO Materials Biocompatibility Matrix can help you to determine if your device needs testing. However, you can reduce the number of tests your device requires, or even eliminate them altogether, if you provide sufficient justifications based on the following types of data:
- Data from previous submissions. If the materials you are using are substantially the same as those previously tested, you may not have to do further testing. If there are any significant changes, such as in the sterilization method, manufacturing process, the nature of patient contact, the chemical composition of materials or materials selection, then confirmatory testing may be required.
- Data from suppliers of the materials being used. If they have conducted their own testing, you will need access to the data.
- Clinical data. For example, there may be predicate devices or devices containing similar materials that have been approved for the market.
- Analytical data. This may be used to affirm low risk.
The FDA guidance I mentioned earlier includes a flowchart to help you decide whether you need to perform biocompatibility testing, which we've reproduced for you here:
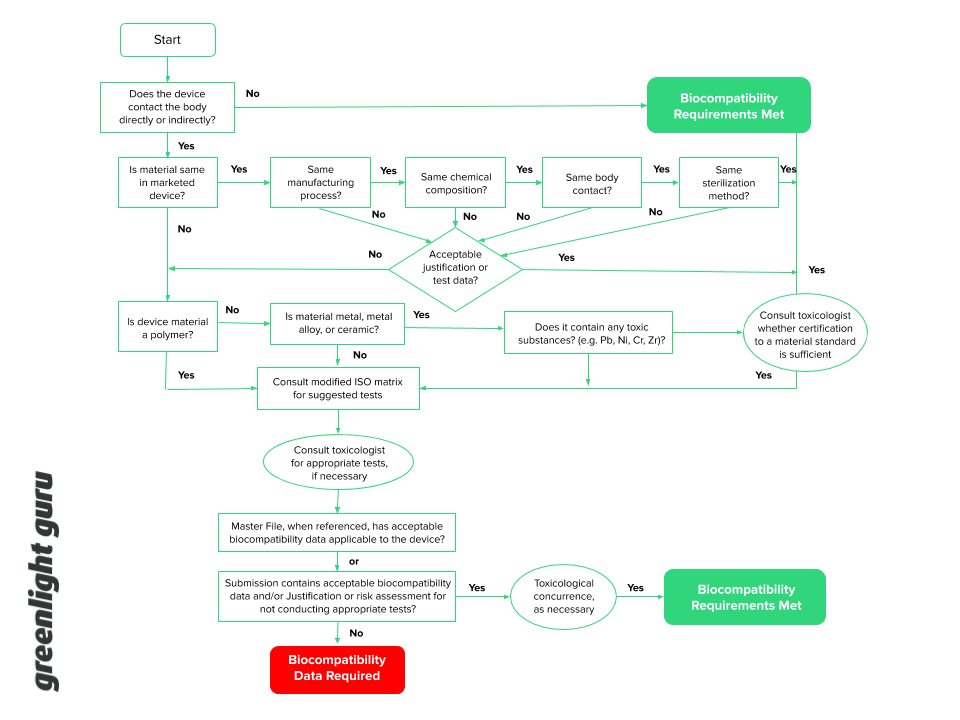
FDA CDRH Biocompatibility Flow Chart
What biocompatibility tests do I need to perform for my medical device?
The exact tests that you’ll need to do vary according to the data and information you have as per the last section here. There are three steps to demonstrating biocompatibility:
-
Develop a Biological Evaluation Plan (BEP). This reviews your device and its materials, identifies potential risks and suggests possible evaluations or testing to address those risks. The BEP can serve as your initial risk assessment and you can also share it with the FDA during a free pre-submission discussion. This is a good idea to use this service and find out if the FDA might require anything further that you haven’t included.
-
Device evaluation and testing. This involves using the tests identified in your BEP. Usually, these will be a combination of in vivo or in vitro biological tests, chemistry tests and toxicological risk assessment, written assessment based upon scientific literature. Pacific Biolabs presents a good summary of the types of tests here.
-
Produce a Biological Evaluation Report (BER). This is where the results of all tests and evaluations are summarized. This is submitted to the FDA along with test results.
Use of biocompatible materials in medical devices
Here’s a question many medical device developers ask: “Is there a list of approved biocompatible materials from the FDA?” The answer to this is no. FDA doesn’t approve materials, they approve medical devices and there are many reasons that a material which is biocompatible in a specific device may not be biocompatible in a different device with a different intended use and indications for use.
What may help is to look at predicate devices—those that are substantially similar to your device or which use the materials you are proposing in a similar way. Data from predicate devices that are already approved can be used as part of your justification for the biocompatibility testing you do (or don’t).
There are some materials for which there are “master files” filed with the FDA, but the FDA does not own those—the company that prepared them does. You might be able to get permission from the file owner to view it, but it’s important to understand that this information is not pre-approved by the FDA.
FREE DOWNLOAD: Click here to get a printable PDF of the biocompatibility evaluation flow chart.
Integrate your biocompatibility testing & risk management into all product development activities with Greenlight Guru Quality
You should always address biocompatibility testing for a new medical device. It’s part of the requirements to ensure that risk is managed and that safety is prioritized for all materials that come in contact with users.
Keep in mind that your risk management process, including biocompatibility testing, should be integrated with the rest of your quality management system (QMS), including design controls. And with Greenlight Guru Quality, that connection comes standard.
Our fully-connected QMS software breaks down information silos within your QMS, ensuring end-to-end traceability. And with a dedicated Risk Management workspace that’s seamlessly integrated with your design controls, you’ll ensure that risk is a living process throughout the entire product lifecycle.
Ready to see for yourself? Then get your free demo of Greenlight Guru today →



