.png?width=800&height=400&name=5%20Tips%20for%20Designing%20a%20Medical%20Device%20Study%20Under%20FDA%20Requirements%20(new).png)
Let’s face it—medical device clinical studies are no walk in the park. There are so many variables to consider, such as patient recruitment, study design, and regulatory requirements—and missing the mark on any one of those could get device manufacturers off on the wrong foot.
So, how do you get started on the right foot, and what are some of the key considerations before moving forward with your clinical study design?
For me, the only way to avoid these bumps in the road is by following best practices in the design of clinical studies. This will not only set manufacturers up for a successful study with fewer constraints and lower costs, it will also ensure sufficient, high-quality, GCP-compliant data that facilitates FDA submissions and boosts market success.
To get a better grip on what those best practices are, we hosted a free webinar to provide you with 5 key tips for the design of clinical studies in both US and EU markets. Here’s a full roundup of the insights shared by Pall Johannesson Co-founder & Managing Director of Greenlight Guru Clinical, and Stephanie Mull, Senior Director of Clinical Operations at Proxima CRO.
Avoid the common pitfalls of clinical study planning and design
Before planning a clinical study, you have to learn to avoid some of the most common clinical issues that befall so many medical device organizations.
-
Avoid paper, go with digital from the start. Companies who choose the digital approach—rather than collecting clinical data on paper—-collect and monitor data much more efficiently. They also save time and resources when it comes to complying with ISO 14155:2020. On top of that, a digital study means you can monitor your clinical data remotely and in real-time.
-
Don’t collect too much data, as it overwhelms teams. We’ve often seen early-stage companies that are embarking on their first clinical study, but also larger companies with a massive budget, ending up collecting more data than they need. In doing so, they greatly increase the workload on clinical staff, e.g., clinical investigators, coordinators, nurses, etc., who might then feel more frustrated and less motivated.
-
Never forget the individual. As device manufacturers, your focus is understandably on your device. You perform pre- and post-market activities where you study how your device is performing on safety and efficacy parameters. However, when you define how you want to run your study, you need to remember that at the center of it all is the patient. Patient Reported Outcome (PRO) data is not very difficult to get as it is data that patients are usually very motivated to provide.
Optimize clinical study resources and reduce costs
Clinical studies can often take hundreds, if not thousands of hours to complete. With that in mind, assembling a dynamic team is essential to a successful study—particularly one that comes in at or under budget.
Optimizing your clinical study is really a process of evaluating your team structure and resourcing needs. What in-house skills do you already have, and which ones will you need to source externally?
-
Executive Oversight and Project Management to manage the trial from beginning to end and ensure successful delivery of that trial within timeline and budget.
-
Site Contracts, Budgets, Payments to manage the contracting and budgeting for sites, as well as to ensure that sites are properly paid.
-
Safety Reporting to monitor and respond to handle adverse event (AE) reporting.
-
Clinical Monitoring & Site Management to monitor data and oversee site management activities.
-
Data Management to build the database, clean the data, and ensure the data is secure and complete.
-
Biostatistics to analyze the data and create your final CSR.
-
Investigational Product to manage your intellectual property (IP), obtain the necessary patent and trademarks, and guide your product portfolio.
-
Medical Writer to compose and manage clinical documentation like your protocol, informed consent, and CSRs.
Another important aspect of resource allocation and planning is to understand your key project deliverables. Below, you’ll see a timeline of the most common milestones during a clinical trial.
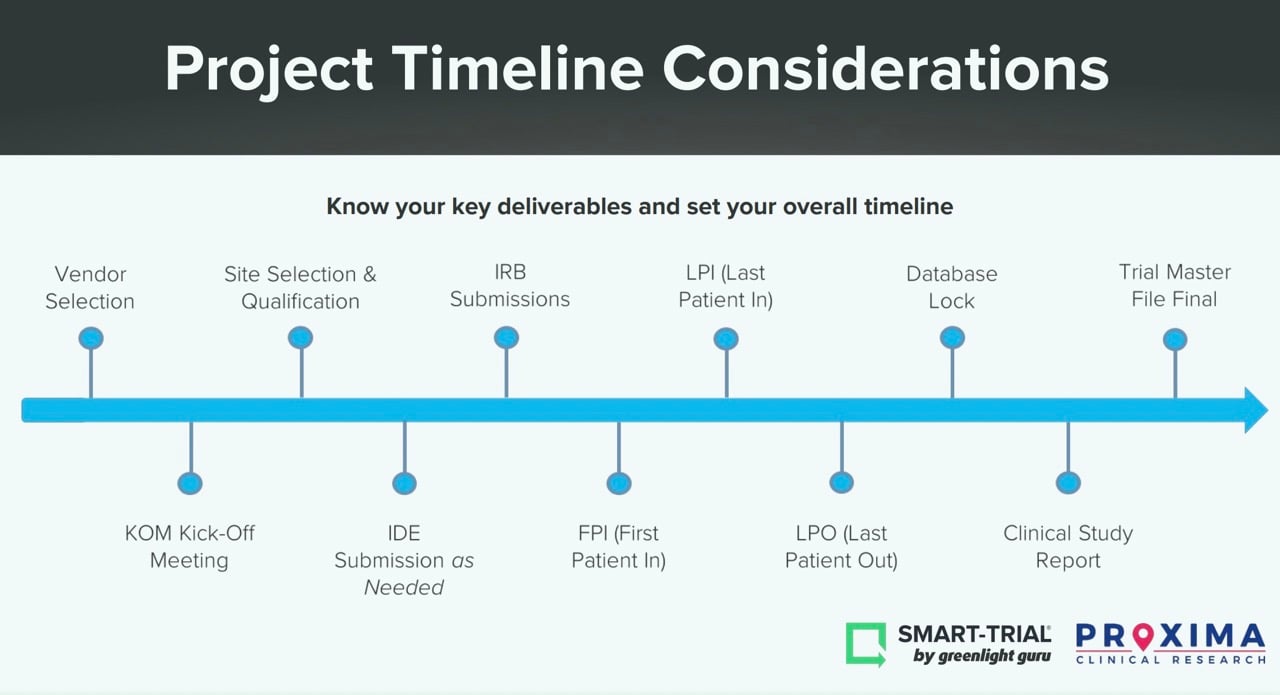
What are your anticipated timelines? What dependencies are there between milestones? For instance, reaching your First Patient-In depends heavily on having a final protocol and site selections in place. Another tip—don’t overestimate the enrollment time. The enrollment process takes a significant amount of time and most sites will rarely deliver those faster than anticipated.
Carefully consider the site selection for your clinical study
There’s no doubt about it—site selection will be a make-or-break factor in the success of your clinical study. Your study site sets the stage for your recruitment, data quality, and overall study costs. That’s why you need to be vetting your potential sites and using an established criteria—not just for the location, but for the investigators who will manage your clinical operations.
The investigator is responsible for the preparation and execution of the protocol for the study, monitoring the safety of the study, the collection and analysis of the data, and reporting the results of the study. With so much at stake, there are a few essential qualifying considerations that must be asked of any site investigator.
-
Experience. The investigator should have relevant experience in conducting clinical studies and have expertise in the relevant therapeutic area.
-
Competing Studies. Investigators who are involved in too many studies may not be able to dedicate sufficient time to the trial, which could impact recruitment and enrollment.
-
Resources and Time. The investigator should have access to adequate resources and time to conduct the trial effectively. This includes having sufficient staff and equipment to conduct the trial and being able to dedicate the necessary time to the study.
In terms of site selection, your criteria should examine the following factors:
-
IRB/Ethics Committee. The site should have an Institutional Review Board (IRB) or ethics committee responsible for ensuring that the study is conducted ethically and in compliance with applicable regulations and guidelines. The IRB ensures that the study is conducted in a manner that respects the rights and welfare of the study participants.
-
Scientific Committee. The site should also have a scientific committee responsible for ensuring that the trial is conducted according to the protocol and that the study results are valid and reliable. The scientific committee ensures that the study is conducted in a scientifically rigorous manner.
-
Site Document Procedures. The site should have procedures in place for document management, including document creation, storage, and retrieval, to ensure that study records are complete, accurate, and secure. Site document procedures ensure that the study records are maintained in a manner that ensures their integrity and security.
-
Contract and Budget. Finally, the site's contract and budget should be carefully reviewed and negotiated to ensure that the site is committed to conducting the trial effectively and efficiently. A well-written contract and budget agreement ensures that the site is committed to the study's success.
Protect patients with important safety considerations
Another important consideration should be the safety profile of your product—and how you plan to manage any safety events that may occur.
For starters, you want to make sure you understand the regulatory reporting requirements for both Serious Adverse Events (SAEs) and Unexpected Adverse Device Effects (UADEs). Investigators and sponsors must report SAEs and UADEs to regulatory authorities and ethics committees within specified timelines. Failure to report these events in a timely and accurate manner could result in serious consequences for patients and the clinical trial.
A Safety Management Plan (SMP) is a comprehensive plan that outlines the safety measures to be taken during a clinical trial. An SMP should include the procedures and policies for managing safety events, risk management, and data monitoring. The SMP should be developed at the beginning of the clinical trial and updated as necessary throughout the trial's duration.
A validated safety database is an essential tool for managing safety data in clinical trials. A validated safety database provides a secure platform for collecting, managing, and analyzing safety data. The database should be designed to capture all safety events, including SAEs and UADEs. The use of a validated safety database ensures that safety data is accurate, complete, and easily accessible.
Lastly, you should keep in mind that in some cases, a Clinical Evaluation Committee (CEC) may be necessary for adjudicating safety events. A CEC is an independent committee responsible for reviewing safety events to determine whether they meet the criteria for SAEs or UADEs.
Produce higher quality clinical evidence with fewer constraints
One final bit of wisdom: you need to remember that your study design will be impacted by how your device is used. In other words, try to design your study so that entering data or giving feedback on the performance of the device is thought of as part of the natural flow of using that device.
This translates to a few tangible and simple principles.
-
Don’t try to put extra strain on the clinicians at the wrong time or introduce extra steps that result in worse data.
-
Keep in mind that not all the sites are the same. There are differences between one clinic and another in terms of what they are used to doing and their work process.
-
Test, test, and test. I cannot stress the importance of testing your study with real users enough. Start testing studies internally with colleagues, try to test them with clinicians if they are involved in collecting the data, and test with patients.
-
Leverage purpose-built tools for your electronic data collection. Remember that medical device clinical studies are built differently, and don’t very well align with the general purpose tools created for the pharmaceutical operation model. Clinical data collection in device studies and clinical operations for devices is conducted around the whole life cycle of a device (from early stage to later stage for market approval, and post-market).
When you choose Greenlight Guru Clinical, you are leveraging the leading clinical data collection toolbox that is purpose-built for the nuances of MedTech clinical trials. Now, designing and optimizing both pre and post-market clinical studies is a thing of beauty. Not only that, but Greenlight Guru Clinical meets the regulatory requirements of the FDA, EU, and most other countries, and ensures compliance out-of-the-box with GCP and ISO 14155:2020.
If you’re ready to start designing the optimal medical device studies, contact us today for a demo of Greenlight Guru Clinical.
ON-DEMAND WEBINAR:
5 Tips for Designing a Clinical Study under US FDA Requirements
5 Tips for Designing a Clinical Study under US FDA Requirements






