
What’s this guide about anyway?
Back in 1998, I started my career as a medical device product development engineer.
At that time the FDA Design Controls regulations were still fairly new -- not only to me -- but the industry in general. ISO 13485 and corresponding design and development requirements for the medical device industry were also very new to the industry in the late 90s.
In those days, we all struggled to understand how and what to do with respect to Design Controls.
As my career progressed, I started to understand the purpose and intent of Design Controls.
But it didn’t happen overnight.
Prior to founding Greenlight Guru, I was fortunate to have played a part in getting 40+ medical devices through regulatory clearance in a variety of different roles.
Since starting Greenlight Guru, a Quality Management Software platform designed specifically and exclusively for the medical device industry, we have been a part of helping dozens and dozens of companies all over the world bring their products to market.
However, for many, Design Controls is still a topic that is as confusing today as it was for me many years ago.
In fact, it is quite common for me to hear negative comments about design controls when I speak to product developers. I assure you that if you have bad feelings about design controls, it is likely because of the processes you are working within; design controls are what we do as prudent product developers.
Design controls demonstrate our medical devices are safe, effective, and meet the indications for use.
With this guide, I plan to share valuable insights to explain what Design Controls are, how to address them, and how they benefit your medical device product development efforts.
Some key questions and concepts that will be covered in this guide include:
- How do design controls apply to simple devices, complex devices, and software as a medical device?
- How can risk management practices be integrated throughout the design and development process?
- Why is design controls traceability so important?
- How do you handle document-based processes for documenting design controls?
You have an idea for a new medical device. Now what?
Congratulations! You have an idea for a new medical device.
Chances are you believe your idea will have a significant impact on the quality of our lives in some way. Chances are your product will help solve some current problem and address unmet needs.
What do you do now?
Throughout the world, there are agencies that govern and regulate medical devices. For example, in the United States, medical devices are regulated by the Food & Drug Administration (FDA). In the EU, there is the European Competent Authority. Canada has Health Canada. Australia the Therapeutics Goods Administration (TGA). And so on.
These regulatory agencies have defined rules and regulations that you and others developing and manufacturing medical devices must follow. And these regulatory agencies have defined rules and regulations about how medical devices are classified and what is required before the products are sold into the marketplace.
Most notably, since you have an idea that you want to develop further, there are regulations established for you to follow during the product development process. These regulations are known as Design Controls.
This may sound confusing and discouraging. I get that.
And I don’t want you to be confused or discouraged.
This is what drove me to create this “Ultimate Guide to Design Controls for Medical Device Companies.”
In this guide, I will share with you the necessary background about Design Controls from a global regulatory perspective.
I will provide you with knowledge and information that arm you with more than just the basics.


Some explanation about a Quality System
Yes, I know -- this guide is supposed to focus on Design Controls. And it does.
I just need to spend a few minutes explaining what a quality system is and how this relates to your medical device product development efforts.
What is a Quality System?
A medical device company has to establish a quality system.
A quality system is a set of processes and procedures you define and implement to describe how your company addresses medical device regulations, including Design Controls.
FDA defines the rules in 21 CFR Part 820. And if you plan to go to market in the U.S., these regulations are required.
Outside the U.S., Europe requires a quality system to be established to meet the medical device regulations (and/or IVD regulations). Many medical device companies choose to implement a quality system and have it certified to ISO 13485:2016 to satisfy EU needs.
Canada -- same thing. The expectation is that a quality system be established. Canada is a little different, requiring you to establish ISO 13485:2016 certification and the Canadian Medical Devices Conformity Assessment System (CMDCAS) (and starting January 1, 2019, a MDSAP - medical device single audit program audit).
The good news is this... FDA 21 CFR Part 820 and ISO 13485 are very similar. In fact, the “new” ISO 13485:2016 is in one-to-one alignment with FDA 21 CFR Part 820.30 regarding Design Controls. This is good news and means you can establish a “one size fits all” quality system, encompassing Design Controls too.
Build your Quality System as you go (For Startups)
For medical device startups, the quality system expectations are that you have all parts and pieces defined and implemented by the time you go to market.
Yes, there are parts of the FDA regulations and ISO requirements that do apply to you, even if you are pre-market.
If you are going through medical device product development, there are at least 4 parts of a quality system that you need to put in place:
- Design Controls
- Risk Management
- Document Control & Records Management
- Supplier Management
The term I use is “bootstrapping a QMS”. I encourage this approach. Early on, you don’t need to spend too much time implementing a robust quality system. You need to be focused on product development.
And as you get closer and closer to going to market, there are software tools, like Greenlight Guru and others, you can use to gradually implement more and more of a QMS.
Just make sure you always keep your quality system in mind from the beginning, so you don’t have to learn how to free yourself from a quality system nightmare down the road.

What are Design Controls?
So far in this ultimate guide, I have spent very little time discussing Design Controls. Yes, this is deliberate.
The stuff I covered so far about understanding medical device product classification and quality systems is very important for you to have some grasp on as you pursue your new medical device idea.
If that information did not discourage you and you are still reading, rest assured.
The rest of this guide is devoted to Design Controls. Really more than just Design Controls.
But first, a quick definition.
Design Controls are a set of FDA-regulated processes that medical device companies must follow to ensure that a device is designed to meet user needs, regulatory requirements, and intended purposes.
I will also share with you why Design Controls even matter and how they will help you during your medical device product development.
Design Controls - Comparing FDA and ISO
It is important that you design and develop a medical device that is safe. FDA, European Commission, Health Canada, and all other regulatory bodies throughout the world will want some assurances that your medical device is safe before you bring the product to market.
And this is really the essence of Design Controls. Proof that you have designed a safe product that meets user needs and requirements.
Technically speaking “Design Controls” is a FDA term and is defined in FDA 21 CFR 820.30 (the 21 CFR stuff is FDA terminology to describe where in the code of federal regulations the topic is addressed).
In ISO 13485 speak, the terminology and intent is similar and covered in section 7.3 Design and Development.
The table below compares the FDA clauses for Design Controls to ISO 13485:2016 clauses regarding Design & Development.

Both FDA Design Controls regulations and ISO 13485 Design & Development requirements expect you to keep documentation and records throughout the product development process.
The Design History File (DHF) is a great place to keep all of your Design Controls “evidence”.
Importance of Traceability
An industry best practice is to construct a traceability matrix to show the linkages and relationships between User Needs, Design Inputs, Design Outputs, Design Verification, and Design Validation.
Building and maintaining your traceability matrix using tools like Excel or Google Docs can be a fairly straightforward task during the very early months of product development.
And as your project progresses, you’ll soon find using these general-purpose tools take days, if not weeks, to properly update and maintain your traceability matrix.
Don’t believe me? Ask an experienced medical device product development project manager about how much time was spent keeping traceability up to date throughout a project. It’s very likely this took 100s of hours per project per year.
This marks a time in your project when switching to a software solution built specifically to meet the unique regulatory needs of a medical device company, like Greenlight Guru, can result in significant gains in time to market while reducing risk.
A Design Controls traceability matrix is vital to product development teams, especially for project managers.
Why?
Traceability shows the relationship and linkages between all of your Design Controls. How do User Needs relate to Design Inputs? How do Design Outputs relate to Design Inputs? How do Design Verifications link to Design Inputs and Design Outputs? How do Design Validations relate to User Needs?
A traceability matrix is an invaluable tool to show a high-level view and the flow of medical device product development from beginning to end.
Best practice product developers have relied on Design Controls traceability for many, many years. And now ISO 13485:2016 also makes traceability a requirement. Here are a couple of citations from ISO 13485:2016 on this topic:
7.1 Planning of Product Realization
- c) required verification, validation, monitoring, measurement, inspection and test, handling, storage, distribution, and traceability activities specific to the product together with the criteria for product acceptance
And
7.3.2 Design and Development Planning
- e) the methods to ensure traceability of design and development outputs to design and development inputs
Incorporate Risk Management
It is an expected best practice that you integrate Design Controls and Risk Management during your medical device product development efforts. For Risk Management, be sure your approach is up-to-date and aligns with ISO 14971.
The Definitive Guide to ISO 14971 Risk Management for Medical Devices
Design Controls are intended to demonstrate that a medical device has been:
- Designed to address the needs of users and patients.
- Designed to meet inputs and requirements.
- Proven to meet applicable standards.
- Meets performance criteria.
Your Design Controls will prove that your medical device is safe for use.
Does this sound like Risk Management?
The intent behind Risk Management is to identify, evaluate, analyze, assess, and mitigate potential product issues.
There is a very strong correlation and relationship between Design Controls and Risk Management.
With Design Controls, you also identify, evaluate, analyze, assess, and mitigate potential product issues.
Design Controls and Risk Management address design, development, and manufacturing of medical devices from slightly different perspectives.
Good Design Controls Reduce Product Risks
If you are thorough with defining and documenting User Needs, Design Inputs, Design Outputs, Design Verification, Design Validation, and Design Reviews, then you will be on the right track towards ensuring your medical device is safe.
Prior to clinical use, you have to know without a doubt that the product is safe and/or determine that the medical benefits outweigh the risks (which should be documented in a risk/benefit analysis).
Embrace this in your own medical device product development efforts.
Realize Design Controls and Risk Management are related.
Realize that your overall goal in medical device product development and manufacturing is to prove and demonstrate that your product meets clinical needs, design inputs, and requirements, and is safe and effective.
Risk Management Is Still Needed Even With Good Design Controls
Having solid Design Controls in place is NOT a substitute for Risk Management. Both are needed.
Realize that Risk Management is just as important (maybe more so) than Design Controls.
Realize that Risk Management is a way to evaluate your product from a different perspective.
Realize that good Risk Management involves a series of tools, when used properly, will drastically improve the quality, safety, and effectiveness of your medical device.
Risk Management & Design Controls Must Be Linked
The best practices of medical device product development have a good flow between Design Controls and Risk Management.
For example, as you identify hazards and hazardous situations, these should “feed” into the Design Controls process in defining User Needs and Design Inputs.
When you evaluate risks, you will need to establish Risk Controls to mitigate and reduce risks. Design Outputs, Design Verifications, and Design Validations become these risk controls.
In fact, using Risk Management as a real tool will help you with Design Verification and Design Validation planning. Let me explain.
Risk Controls are used to help identify ways to reduce the risks. Your Risk Controls should align with and include Design Verification and Design Validation activities.
Are you starting to see how closely related Risk Management and Design Controls should be?
Product Development versus Design Controls
I’d like to spend a few minutes discussing product development versus Design Controls. Is there a difference? Or are these terms synonymous?
The short answers to these questions depend on who you ask.
And I’m telling you the answers really don’t matter all that much.
When you have an idea for a medical device that you want to turn into an actual product, you will follow some sort of product development process. And I highly encourage you to map your product development process and WRITE IT DOWN.
As an example, I like to define the product development process in major phases:
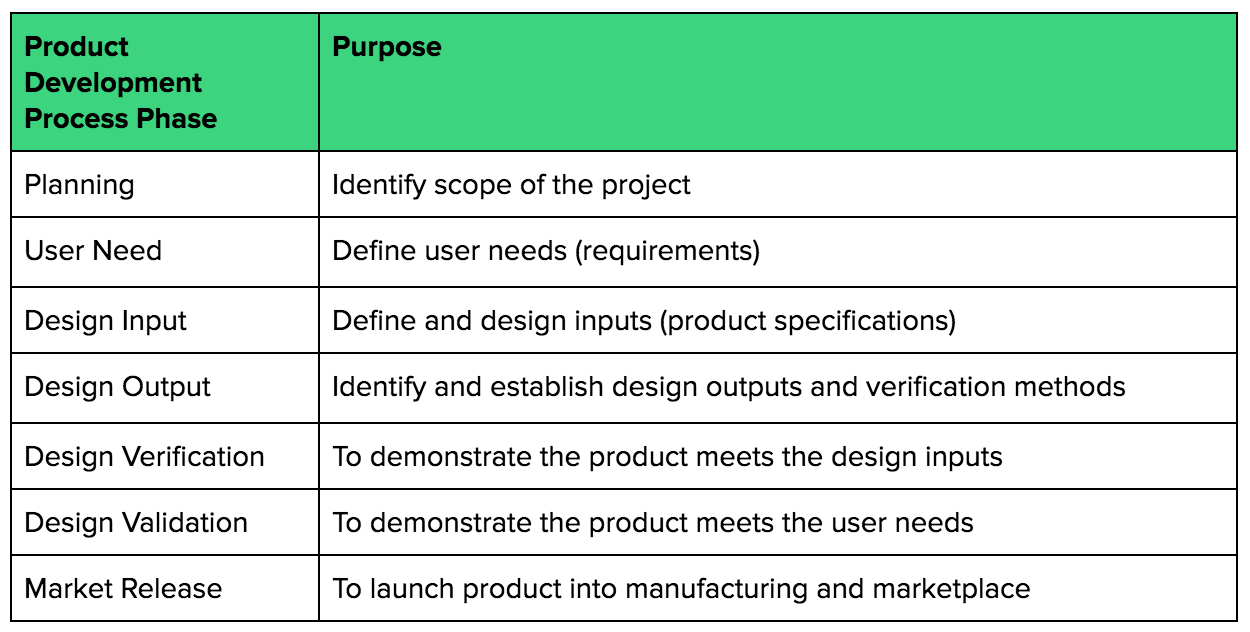
During product development, you will construct some prototypes, do some testing, get some feedback, etc. Always trying to get to the next step towards launching your medical device.
As noted above, Design Controls are all about ensuring the medical device you are developing is safe.
All about making sure you have proven your medical device meets the requirements you define.
All about making sure you have proven your medical device meets the user needs and product’s intended use.
All about making sure you will be able to manufacture your medical device that you designed and developed.
But since I posed the question about product development versus Design Controls, I’ll give you my opinion.
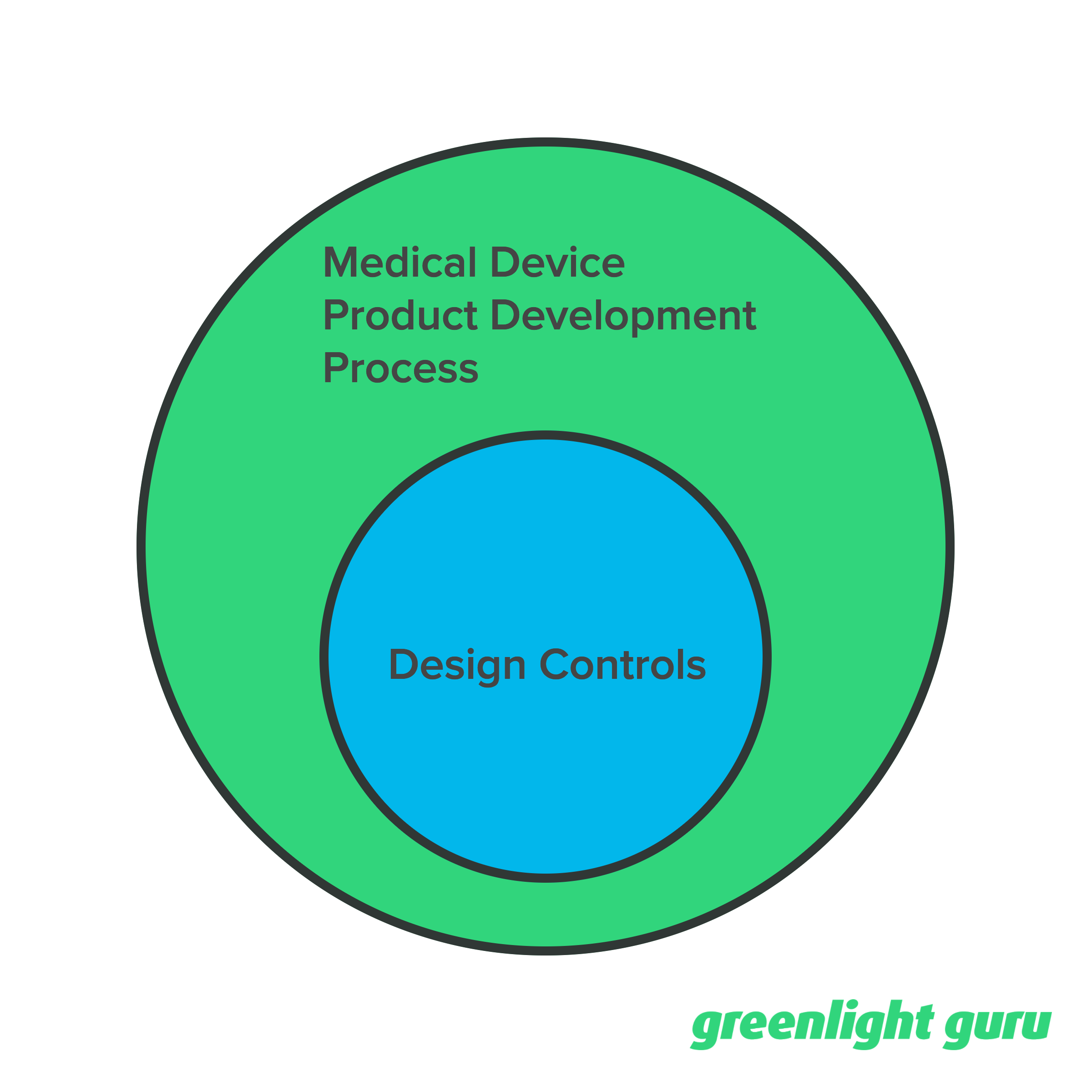
It’s my opinion that Design Controls fits within the broader medical device product development process.
Product development includes much, much more than just Design Controls. Things like budget, timeline, business development, marketing, sales, and so on.
Product Development Process Methodology / Approach
I often hear that the Design Controls requirements are often restrictive and require a phase / stage-based approach (some refer to this as “waterfall”) for the product development process.
Let me set the record straight.
FDA does not mandate a waterfall approach for medical device product development and capturing Design Controls. In fact the FDA Design Control Guidance published in 1997 is very clear about the iterative nature during device development.
I bring this up largely because of the current popularity of “agile” methodology for product development, especially with complex devices and software as a medical device (SaMD).
For example, the image below is the “V-model” described in IEC 62304, a standard that describes software life cycle product development processes.
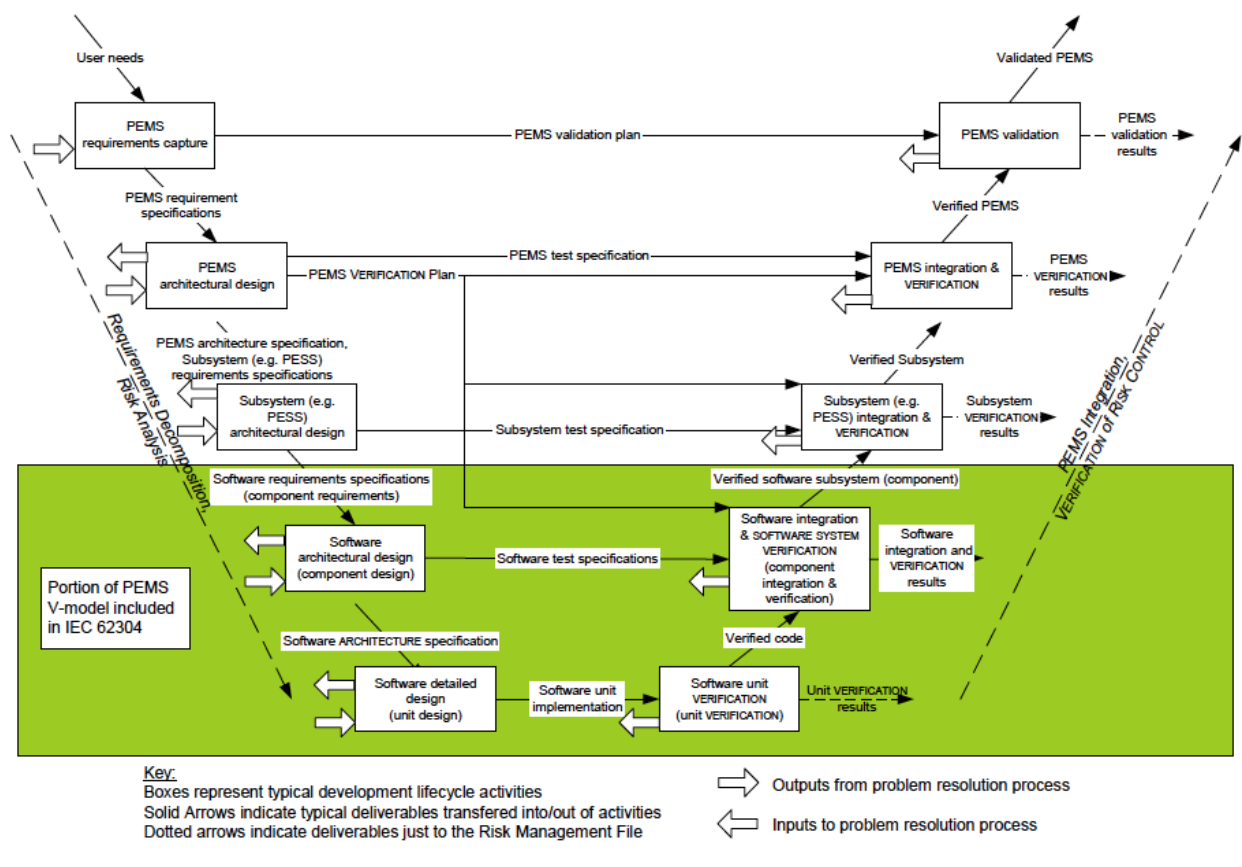
IEC 62304 Medical device software – Software life cycle processes
Figure C.2 – Software as part of the V-model
Note, the v-model describes a product development methodology and approach.
Regardless of product development methodologies and approaches used, demonstrating Design Controls as defined by FDA and ISO 13485 are still a requirement.

Maintaining a Design History File
What is a Design History File (DHF)? When you read FDA Design Controls regulations, the part about DHF is mentioned towards the end. And ISO 13485 doesn’t officially talk about a DHF.
I want to cover DHF now so that you understand what it is and why it matters.
A Design History File is, simply put, the place where you keep all your Design Controls.
A DHF should be organized and accessible to the entire project team.
A DHF is the place where you will show the linkages and relationships between all the Design Controls.
Demonstrating traceability of all your Design Controls throughout the product development process is expected and required.
Whether you use a paper-based approach, general purpose tools, or a software solution designed specifically with Design Controls traceability in mind like I mentioned before, know that the responsibility to prove this is yours.
Keep the DHF separate from project file
My comments about product development versus Design Controls may seem somewhat trivial.
Except that it isn’t.
And the DHF is my main reason for saying so.
Yes, Design Controls and the DHF where these documents and records are maintained are important.
The DHF is the central hub for all the things medical device regulators care about.
Which is why it is so important to keep your Design History File as a stand-alone thing.
Going through medical device product development, you seldom consider how to maintain documentation and why it even matters.
And after you launch your medical device, the DHF is important in the event the project is ever audited by the FDA or others.
The last thing you want to do when finishing up a project is to spend weeks going back organizing documentation and records, including the Design History File.
I’m telling you that if you do not keep things shipshape as you go, you will not go back to clean it up.
This is why I recommend building your Design Controls traceability matrix early and keeping it up to date as you go, whether you use purpose built software or not. With this approach, you won’t stress about the possibility of being audited because your DHF will always be up to date and audit ready.
.png)
Click here to learn more about Greenlight Guru's new Multi-Level Design Control Management software.
Ensure the DHF is organized and accessible
Ensuring Design Controls documentation and records are stored in a DHF is very important.
Keeping the DHF in a place where your entire product development project team can access it is also important. Why? Throughout product development, your team will be contributing valuable content to include in the DHF, and accessing this same content to conduct product development.
Be sure you are deliberate in how you plan to keep the DHF organized and accessible.
Do you plan to use a paper-based approach? If so, be sure you are aware of the challenges paper systems pose.
Do you plan to store electronic records on a company server or some type of electronic document management system? There are also challenges with this approach as well.
Once upon a time, prior to founding Greenlight Guru, I often relied heavily on paper-based approaches, general purpose tools, and server folder trees to manage DHFs. And every time, I was faced with the following:
- Have missing documents and records. Flat out. I promise. There will be critical must have documents you need as objective evidence which will be lost, never to be found.
- Documents and records will be out of sync. Managing document revisions in a paper world is an absolute nightmare. Making sure the correct revision is kept and maintained everywhere is a tall task.
- Must have review and approval signatures on documents will be missing. With paper, you need to have actual “wet” signatures on paper. I guarantee that not all documents will have all approval signatures when you use paper.
Do your homework. Choose your solution wisely.

Why capturing User Needs sets the stage
Remember when you had the idea for your new medical device, you came up with this wonderful product because you want to address unmet clinical needs. There are two terms that you should understand: intended use and indications for use.
Intended Use is the general purpose of the medical device or its function (what you “claim” the medical device does)
Indications for Use describe the disease or condition the medical device will diagnose, treat, prevent, cure, or mitigate, including a description of the target patient population
First, intended use is EXACTLY what your product is used for. Don’t focus on what it COULD be used for. Rather, define exactly what it is in as few words as possible.
Second, your indication of use statement are the precise situations and reasons where and why you would use this device. Again, being very clear and definitive here is extremely important.
“They think they understand what intended use means, but they really don’t…Intended use is all about what we say this device is to be used for, and indications for use is under what circumstances or under what conditions you would use that particular product.” – Mike Drues
All of this relates to the User Needs for your medical device and are part of Design Controls.
Now you can do a lot of digging to try to get a better understanding of what User Needs really are. FDA doesn’t really define User Needs in the 820.30 regulations per se. User Needs are shown kicking off the Design Controls process in the classic “waterfall diagram”
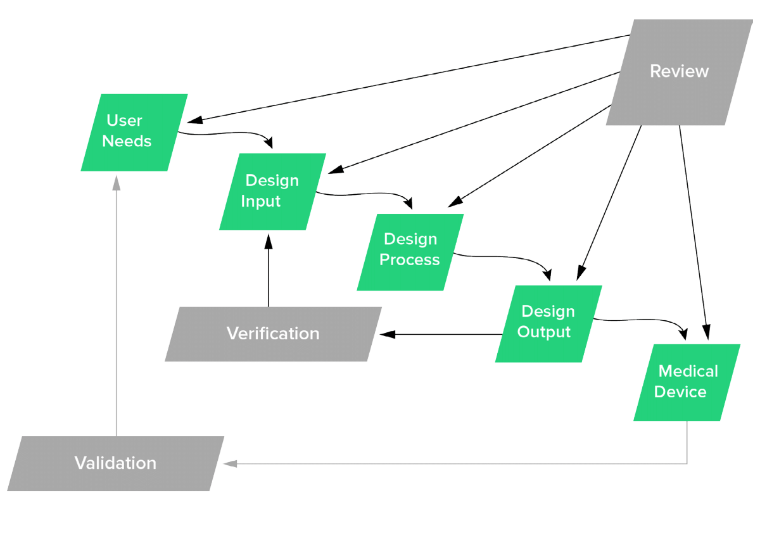
Classic Design Controls waterfall diagram originally published by Health Canada & published in FDA Design Controls Guidance
The classic Design Controls diagram should be viewed as a means to describe the relationship between Design Controls elements. As discussed earlier, this does not mean your medical device product development methodology has to be “waterfall” in nature.
The best place, aside from this guide, for you to get some insights about what User Needs are is the FDA Design Controls Guidance.
And when you read the FDA guidance, just about every reference involving User Needs is tied directly to Design Validation. Validation proves your medical device meets User Needs and intended uses.
Documenting User Needs
Okay, so let me get into documenting User Needs. When documenting User Needs, think about your medical device product idea.
User needs describe how your medical device is going to be used. User needs help establish the framework for your medical device product design. Intended use describes the clinical issue your product addresses. Indications for Use pertain to clinical applications use, environment, and end user.
Here are some questions to help you better understand User Needs.
- What do you want it to do?
- Who is going to use it?
- When will it be used?
- What important features and attributes should be considered?
- How will the user and patient interact with the device?
- What type of procedures will the device be used?
- What type of environment will the device be used?
- When will the device be used?
- Is the device used one time or over and over?
- What other products will the device interact and interface with?
There likely are dozens and dozens more questions you need to ask.
And answer! Document your responses to all the questions.
Your responses become the information you should consider as the User Needs for your medical device idea, and included as part of your Design Controls.
Looking again at the waterfall diagram, you see that the proposed next major step after User Needs is Design Inputs. Logically, User Needs are the lead into Design Inputs.
And many medical device product development professionals will share with you how important Design Inputs are to the success of a new device.
Do not be too worried about whether or not your User Needs are exactly correct. Do not get too hung up on the format and wording.
Do realize that one day in the future, you will need to demonstrate that the product you develop addresses the User Needs you define.
The traceability matrix described earlier in this guide starts with the User Needs. User Needs feed into Design Inputs. Design Validation proves User Needs are met.

The purpose behind Design & Development Planning
In my experience, the topic of Design & Development Planning is very misunderstood. When most medical device product developers think about Design & Development Planning, the first thing that comes to mind is this:
Project schedule.
Since there is confusion about this, let me share what FDA and ISO state about Design & Development Planning.
FDA 21 CFR 820.30(b) Design & Development Planning: Each manufacturer shall establish and maintain plans that describe or reference the design and development activities and define responsibility for implementation. The plans shall identify and describe the interfaces with different groups or activities that provide, or result in, input to the design and development process. The plans shall be reviewed, updated, and approved as design and development evolves.
ISO 13485:2016 7.3.2 Design and development planning
The organization shall plan and control the design and development of product. As appropriate, design and development planning documents shall be maintained and updated as the design and development progresses.
During design and development planning, the organization shall document:
- a) the design and development stages;
- b) the review(s) needed at each design and development stage;
- c) the verification, validation, and design transfer activities that are appropriate at each design and development stage;
- d) the responsibilities and authorities for design and development;
- e) the methods to ensure traceability of design and development outputs to design and
development inputs;
- f) the resources needed, including necessary competence of personnel.
Did you come across anything that suggests a project schedule? Or timeline?
Your product development schedule is definitely important. Don’t mishear me.
Remember where I shared some thoughts about product development versus Design Controls earlier in this guide?
Project schedule versus Design & Development Planning is in the same vein. Project schedule is a product development thing. Yet there is also a definite relationship with Design & Development Planning.
What goes into a Design & Development Plan?
Let me pick out the important aspects about Design & Development Planning from what FDA and ISO state about the topic:
- Identify Design & Development stages and activities
- Define responsibility for Design & Development activities
- Define the resources required; this includes project team members, as well as vendors
- Plans should evolve throughout Design & Development
A Design & Development Plan describes all the Design Controls, including when Design Reviews are expected. The plan describes who the team members are, including roles and responsibilities. The plan is not a “one and done” and needs to be revisited and updated throughout the project.
To help, here is a table listing the types of deliverables that should be expected and defined as part of your Design & Development Plan.
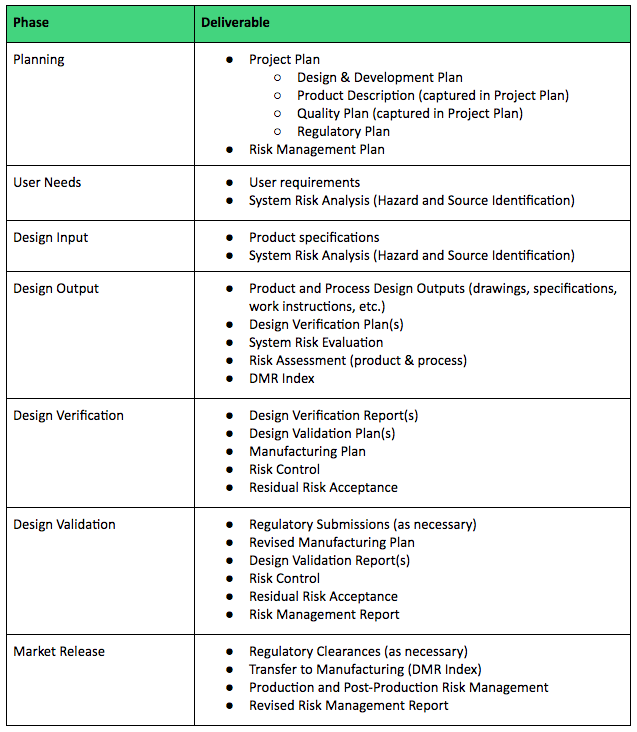


Understanding the role and importance of Design Reviews
Design Reviews are moments in time for you to evaluate the progress of your medical device project as it progresses through development.
During Design Reviews, you and the project team are formally reviewing -- and agreeing to -- Design Controls. This means your team will review and agree to your User Needs, Design Inputs, Design Outputs, Design Verifications, and Design Validations.
Design Reviews are also a time in a project to bring the team together in order to scrutinize and evaluate the state of the medical device and ensure that what needs to be done has been addressed.
How many? Who is involved?
How many Design Reviews does your medical device product development project need? This is a good question to ask. I do not have the answer for your project.
Here’s what I can tell you about Design Reviews.
How many Design Reviews to have depends on numerous factors. Factors like what the product is and who the players are need to be considered when determining the timing of Design Reviews.
The expectation is that your Design Review include the right functions (engineering, marketing, manufacturing, etc.). Also, you need to include a resource who is independent.
When should Design Reviews happen?
All Design Controls must be included as part of a Design Review. As noted, I cannot for certain tell you exactly when to have Design Reviews. If you press me to provide a definitive response to this question, I will defer to what is suggested by the Design Controls waterfall diagram.
I will advise you to have a Design Review when User Needs are drafted.
I will advise you to have a Design Review after Design Inputs are established.
And after Design Outputs. After Design Verification. After Design Validation...and before going to full production.
But if you are following an agile product development process, this approach may not be ideal.
Design Reviews - the absolutes
Okay, let me be more helpful regarding Design Reviews.
First, you must have a record to prove that all Design Controls have been included as part of a Design Review.
Let me provide you with a few “rules” when it comes to Design Review:
- Design Verification can not happen until Design Outputs and Design Inputs are done.
- Design Validation can not happen until User Needs are done.
- Design Transfer can not happen until Design Outputs are done.

Becoming a Design Input artist
Ah, Design Inputs. It is important for you to understand Design Inputs and how critical these are to the entire success of your medical device project.
Design Inputs, in my opinion, are the most important Design Controls.
Keep in mind that throughout medical device product development, the terminology used is grossly inconsistent and highly dependent on who is talking.
What I mean by this is that very few use companies seem to adopt the terminology as described by FDA and ISO. Most have adapted their own lexicon when it comes to medical device product development.
Nowhere is this more true than on the topic of Design Inputs.
Keep this in mind as you define Design Inputs and work with others who will contribute.
Here is a non-inclusive list of terms that are often used to mean “Design Inputs”:
- Design Inputs
- Design & Development Inputs
- Design Input Requirements
- Requirements
- Design Requirements
- Product Requirements
- Requirements Specifications
Why does this matter?
Well, when there is confusion regarding terminology, this can cause miscommunications with your project team. And realize that at some point in time, the Design Controls information you are documenting will be subject to FDA inspections and/or ISO audits. Your terminology will also need to be explained to these auditors.
It can be very confusing. And because of this, I highly encourage you to make sure that you and your team members speak the same language when it comes to Design Controls -- especially Design Inputs.
What are Design Inputs?
Design Inputs define all the performance criteria, requirements, and features of your medical device product.
There are numerous resources that feed into Design Inputs. From a Design Controls perspective, the primary resource that feeds Design Inputs are the User Needs.
Often times User Needs are broad statements that may be difficult to measure. User Needs include words like “easy”, “better”, “simple” -- subjective, abstract concepts. And this is okay. I encourage having User Needs defined in this way.
- Design Inputs, though, should not be subjective, abstract concepts.
- Design Inputs should be objective. Measurable.
- Think of User Needs as the precursor to Design Inputs. (And show how these are linked in a traceability matrix.)
- Do not rely solely on User Needs as the only resource for Design Inputs.
There are things like industry standards, regulations, and competitive products that you should also consider when it comes to defining Design Inputs.
Plus, unless you are tackling medical device product development solo, you have other people working with you who should help define Design Inputs.
Is it necessary to establish a hierarchy of Design Inputs?
How complicated is your medical device? Does it include mechanical, hardware, and software? Are there multiple sub-systems and components? What if your product is SaMD?
If the architecture of your medical device “system” is somewhat complicated in nature, consisting of multiple sub-systems, it may be beneficial to establish a hierarchy for your product.
.png)
Click here to view our Multi-Level Design Control Software.
Doing so will allow you to better define Design Inputs on a per sub-system/component basis. Doing so will allow your project team resources to better contribute, communicate, and stay focused on the parts of your device that apply to them.
For example, keeping your Design Inputs organized in this fashion will allow mechanical engineers to keep focused on mechanical issues.
Think about Design Verification
I know I have not shared what Design Verification is just yet in this guide. Let me give you a preview: Design Verification proves you designed the medical device correctly.
Because of this, you should definitely consider how you plan to verify your product design at the time when you are documenting Design Inputs.
Considering how you will verify will have a profound impact on the content of Design Inputs. You should definitely put thought into how you will prove your medical device will be verified when you define Design Inputs.
Taking Design Verification into consideration when defining Design Inputs will also help your product development efforts. This approach will also help you plan and understand potential Design Verification activities. This is meaningful because Design Verification tasks can be very expensive and take significant amounts of time to complete.
Design Inputs become a roadmap for the product developer
Design Inputs describe everything that is important and required about your medical device.
It is 100% okay that Design Inputs iterate and evolve throughout the product development process. It is 100% okay to use placeholders and language such as “to be determined” when first drafting Design Inputs. In fact, this is really the essence of good design.
Capture as much as you can as early as you can. Identify the items that require more research and investigation. Work towards defining clear, objective, measurable Design Inputs.
Why?
Design Inputs become a roadmap, or set of “directions”, that a medical device product developer uses to design and develop a product.
Design Inputs provide the important criteria that must be included in the design of the actual medical device.

Defining how to make your medical device through Design Outputs
Let’s check in a bit on what Design Controls I’ve covered with you so far.
I shared what a DHF is and why is matters. I explained Design & Development Planning. And importance of Design Reviews.
I talked about User Needs. About how User Needs are the precursor to Design Inputs.
Remember Design Inputs are the roadmap used to design and develop your medical device.
Let me spend a few minutes talking to you about Design Outputs being a recipe for your eventual product and how these contribute to figuring out how to actually design and develop your medical device.
Design Outputs are the recipe
As you get into the hands-on, nitty-gritty design and development, you start to identify all the parts and pieces that are required for your medical device.
What sub-systems and components go into your medical device? How are these components put together?
What I’m trying to say is this: Your medical device is comprised of a number of materials, components, sub-assemblies, and so on. And each of these parts need to be defined and documented.
Things like drawings, specifications, and instructions.
Think of Design Outputs as the recipe for making your medical device. Design Outputs are the documents you would give to someone to assemble your product.
As you establish the Design Outputs, you need to show how these relate and link to the Design Inputs. Do so with a Design Controls traceability matrix (as discussed earlier).
You may think this can be done in Excel quite easily early on in product development but as the project nears market release, these relationships will become increasingly complex.
Keeping a traceability matrix up to date will require several non-value add hours of your time to maintain and update. (This is why Greenlight Guru Design Controls traceability matrix is such a valuable asset.)
Design Outputs are the preliminary Device Master Record
Yes, this guide is about Design Controls. At the same time, it is important how various Design Controls have an influence and impact after you launch your medical device.
Design Outputs have a huge impact on the future state of your medical device. Fast forward with me for a moment.
When you have transferred from product development into manufacturing, you need to establish a Device Master Record for each product.
Device Master Record (DMR) is defined by FDA as:
a compilation of records containing the procedures and specifications for a finished device
A DMR is the recipe.
Just like Design Outputs.

Proving you designed correctly via Design Verification
Let me take you back a couple of topics. Think (or look) back at Design Inputs. I expressed that there is a strong relationship with Design Inputs and Design Verification. I encouraged you to think about how you are going to conduct Design Verification when you define Design Inputs.
When you get to Design Verification, your goal is to prove your Design Outputs meet the Design Inputs. Design Verification is all about demonstrating you correctly designed your medical device.
If you have been keeping your Design Controls traceability matrix up to date, you have a clear picture of the relationship between your Design Inputs and Design Outputs. Having this clear picture is very helpful when it comes to conducting Design Verification.
So how do you conduct Design Verification?
Testing, Inspection, Analysis
When you ask an experienced medical device product developer to describe Design Verification, it’s likely you will hear this described as testing.
“Yes, testing is a very common method used to conduct Design Verification.”
Remember, Design Verification is about proving Design Outputs meet Design Inputs.
Yes, testing is a completely valid way to prove this.
Although testing is not the only way to conduct Design Verification. You might also be able to employ inspection and analysis as acceptable methods for Design Verification.
- Plan your Design Verification before you conduct tests, inspections, and analyses.
- Identify what you are going to do before you do it. Determine the best ways to prove Design Outputs meet Design Inputs.
- And then do it! Complete your Design Verification activities, and document results.
- Update your traceability matrix to show how Design Verification proves your Design Outputs meet Design Inputs.

Proving you have the correct device via Design Validation
Design Verification is about proving you designed your medical device correctly (e.g.: the Design Output meets the Design Input requirements).
Design Validation is about proving you designed the correct medical device.
Please understand the distinction. To do so, it helps to know that Design Validation demonstrates that your medical device meets User Needs and its intended uses.
Design Validation is the time when you revisit these User Needs. Validation is when you demonstrate that the User Needs are met.
Okay, not you per se. But actually end-users. Yes, that’s right -- end users should be involved with Design Validation.
This is the whole intent and purpose: prove your medical device meets the needs of the end user.
Design Transfer begins
Design Validation most definitely involves evaluation of products. And because of this, the process of transferring a medical device from product development to production (often referred to as “Design Transfer”) begins during Design Validation.
In order to validate the design of your medical device, you need to build products. These products are to be built using production equivalent documentation and processes.
So all those Design Outputs (the preliminary DMR) need to be converted to the production environment and used by production personnel to assemble devices.
These production units (maybe the first) are then put in the hands of end users.
Have a plan of attack
If you remember the discussion about User Needs, I shared that sometimes User Needs have a tendency to be a little ambiguous, using words like “easy” and “better”. With Design Validation, you need to figure out how to prove that your product accomplishes this.
You need to establish a Design Validation plan. This plan needs to identify how many end users, what type of testing is required, and so on.
The end goal of Design Validation is to have objective, documented evidence.
Completing Design Validation is an important step in closing the loop on your traceability matrix too. The traceability matrix started with User Needs and ends with Design Validation.

Completing Design Transfer
As I noted, the Design Transfer process actually starts earlier in your project. It begins when you build products for Design Validation.
I also shared how Design Outputs are the preliminary Device Master Record. And the DMR is used by production to make medical devices.
Once you complete Design Validation, you are anxious to officially wrap up product development.
It’s time for you to have a final Design Review. This one may be a doozy (and may be a good idea to break it up in chunks).
The goal of this Design Transfer Design Review is to make sure everything that is needed for production is ready and done.
Completing Design Transfer signifies your medical device is ready to exit product development and officially enter into production.
This is significant because up until this point the “control” of the medical device has been the responsibility of the project team. Once Design Transfer occurs, the control shifts to production resources.
Button up the DHF
I hope you are keeping your Design History File up to date and current as you go through the medical device product development process. This is the best option by far!
Nothing is worse than getting to the Design Transfer stage and discovering that the DHF needs work. It happens. And if your DHF needs work, take the time and do it.
Don’t fall into the trap of saying that you will get to it later.
You won’t.
I’ve shared throughout this guide tips and pointers regarding the need to create and maintain a Design Controls traceability matrix. Keeping your traceability matrix current and up to date makes Design Transfer a much simpler process.
After you complete Design Transfer, the DHF is the ultimate record proving you have satisfied Design Controls.
And your DHF might be reviewed, audited, and/or inspected one day. Keep that in mind.
Preparing regulatory submissions
I want to spend a few moments now talking a bit about regulatory submissions and the relationship with Design Controls.
Generally speaking, as you successfully finish Design Verification, this means you are approaching a time when you can prepare a regulatory submission.
Specifically, this is the time in your medical device project when you should put together your FDA 510(k) submission (provided this is your path to market clearance).
If you plan to conduct a human clinical investigation in the U.S., successful completion of Design Verifications also marks the time when you should put together a FDA investigational device exemption (IDE) submission. (An IDE is likely for FDA PMA devices or could be of interest for clinical investigation before receiving 510(k) clearance.)
FDA PMA devices will be ready for FDA review upon completion of Design Validation.
Outside the U.S. regulatory submissions and files, such as CE Mark Technical Files, are provided to regulatory bodies upon completion of Design Validation, during Design Transfer.

Launch
After you complete your Design Transfer Design Review, you are almost ready for market launch. You also need to ensure that you have all the necessary regulatory clearances.
Then, at least from a Design Controls perspective, it’s time to sell your medical device.


Bringing it all together
Design Controls should be a systematic way to demonstrate the progress of your medical device product development efforts.
In this guide, I have shared with you what is expected for Design History File, User Needs, Design & Development Planning, Design Reviews, Design Inputs, Design Outputs, Design Verification, Design Validation, and Design Transfer.
I hope this guide helps answer all your questions about Design Controls (let me know in the comments!) -- even the questions you didn’t know you had.
Medical device product development should follow a structured, methodical approach with traceability throughout. But we all know nothing always goes according to plan.
That’s why in addition to this guide, we’ve also created Greenlight Guru, an easy-to-use, cloud-based software platform specifically to help medical device companies get to market faster by better managing their Design Controls.
Looking for a design control solution to help you bring safer medical devices to market faster with less risk? Click here to take a quick tour of Greenlight Guru's Medical Device QMS software →
The Ultimate Guide To Design Controls for Medical Device Companies


%20Design%20Controls.png?width=250&height=324&name=(cover)%20Design%20Controls.png)







