
Classification is one of the first hurdles you face when developing software as a medical device (SaMD). Your risk class determines your regulatory pathway, your documentation requirements, and in many cases, the timeline and cost of bringing your product to market.
What makes SaMD classification particularly complex is that there's more than one classification to understand. Depending on where you plan to market your product and which standards you're working with, you may need to work through multiple types of risk and software safety classifications which do not map neatly onto each other.
In this article, we'll break down all the different classifications and requirements by region to help simplify your SaMD classification.
BONUS RESOURCE: Click here to download your free Cybersecurity Gap Assessment Checklist!
FDA SaMD classification in the US
Because software as a medical device is still a medical device, in the United States, the FDA classifies SaMD the same way it classifies traditional medical devices: into Class I, Class II, and Class III, based on risk.
-
Class I devices present the lowest risk and are subject to general controls.
-
Class II devices carry moderate risk and typically require a 510(k) premarket notification.
-
Class III devices present the highest risk and require premarket approval (PMA).
In recent guidance documents, the FDA has begun using the term "device software function" (DSF) rather than SaMD. A DSF is defined as any software function that meets the device definition under the FD&C Act. The terminology has shifted, but the underlying regulatory obligations have not.
One area where SaMDs (or DSFs) differ from traditional hardware devices is in the documentation requirements for premarket submissions. The FDA's guidance on the content of premarket submissions for device software functions describes two levels of documentation: enhanced and basic.
-
Enhanced documentation is required when a failure or flaw in the device software function could present a hazardous situation with a probable risk of death or serious injury, assessed before risk control measures are applied.
-
Basic documentation applies in all other cases.
Importantly, documentation level does not correspond directly with risk class. A Class III device will almost certainly require enhanced documentation, but a Class I or Class II device might require it too, depending on the nature of its software functions. Manufacturers are expected to make this determination individually for each device. For more on what those submissions need to contain, see our post on SaMD software documentation.
Clinical decision support software and FDA oversight
There's one more classification we should talk about when it comes to software in the medical device industry. This type of product is known as clinical decision support (CDS) software, and it may or may not be regulated as a medical device depending on whether it meets certain criteria.
The 21st Century Cures Act amended the Federal Food, Drug, and Cosmetics Act to add section 520(o), which excludes certain software functions from the definition of device in the FD&C Act if they meet four criteria.
The FDA recently put out a guidance document on clinical decision support software that addresses CDS and their interpretation. To qualify for the exclusion from the definition of device, a CDS function must meet all four of the following criteria:
-
It must not acquire, process, or analyze medical images or signals from diagnostic devices.
-
It must display, analyze, or print medical information about a patient.
-
It must support or provide recommendations to a healthcare professional about prevention, diagnosis, or treatment.
-
It must enable that professional to independently review the basis for those recommendations, rather than rely on them as a primary driver of clinical decisions.
If a software function meets all four criteria, it is considered "Non-Device CDS" and falls outside FDA's regulatory oversight. If it fails to meet any one of them, then it remains a regulated device software function.
For SaMD manufacturers, this guidance is a practical tool for scope determination. Many AI and algorithm-driven products sit close to this line, and the guidance includes an extensive library of examples covering both device and non-device functions. If your software is intended to support clinical decisions, working through these criteria early can help you determine whether you are building a regulated SaMD or a non-device tool, and avoid discovering the answer during a premarket submission.
EU MDR classification and Rule 11
In the European Union, SaMD is regulated under the EU MDR (or EU IVDR for in vitro diagnostics). However, the EU does not use the term SaMD. Instead, it refers to "medical device software" or MDSW. The risk classification system mirrors that of traditional devices: Class I, Class IIa, Class IIb, and Class III.
What makes the EU system distinctive for software is Rule 11, found in Annex VIII of EU MDR. Rule 11 provides the specific classification logic for medical device software and is worth understanding in detail.
-
Under Rule 11, software intended to provide information used for diagnosis or therapeutic decisions is classified as Class IIa by default.
-
If those decisions could cause death or irreversible deterioration of health, the software is Class III.
-
If they could cause serious deterioration of health or require surgical intervention, it is Class IIb.
-
Software intended to monitor physiological processes is Class IIa, unless it monitors vital parameters where dangerous variations could create immediate danger to the patient, in which case it is Class IIb. All other software is Class I.
In practice, this means that most medical device software will be classified as at least Class IIa under Rule 11. The European Commission's MDCG 2019-11 and MDCG 2021-24 guidance documents provide further elaboration on how Rule 11 should be applied and are essential reading for any SaMD manufacturer planning to enter the EU market. We break both down in our post on MDCG 2019-11.
IMDRF SaMD categorization
The International Medical Device Regulators Forum (IMDFR) has also developed its own framework for risk categorization, with four possible categories. While the IMDRF is not a regulatory body, it does consist of representatives from many of the largest medical device markets in the world, including the US, Europe, Canada, Australia, Japan, and Brazil, among others. The IMDRF works to accelerate international regulatory convergence, and their working groups have a significant impact on the direction of regulations around the world.
The IMDRF SaMD classification system is two-dimensional. One axis considers the significance of the information the SaMD provides, meaning whether it is used to treat or diagnose, drive clinical management decisions, or inform clinical management. The other axis considers the severity of the condition the software is intended to address, ranging from critical to serious to non-serious situations.
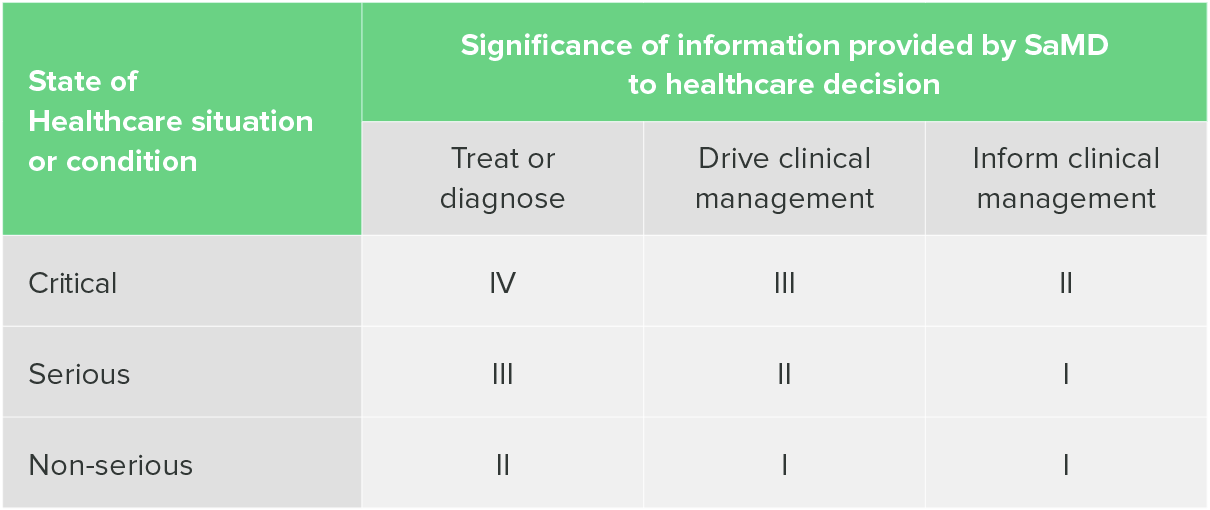
The IMDRF categorization is not widely used as a standalone classification system. Where it does prove useful is in EU compliance, because MDCG 2019-11 includes a table that maps IMDRF categories to EU risk classes.
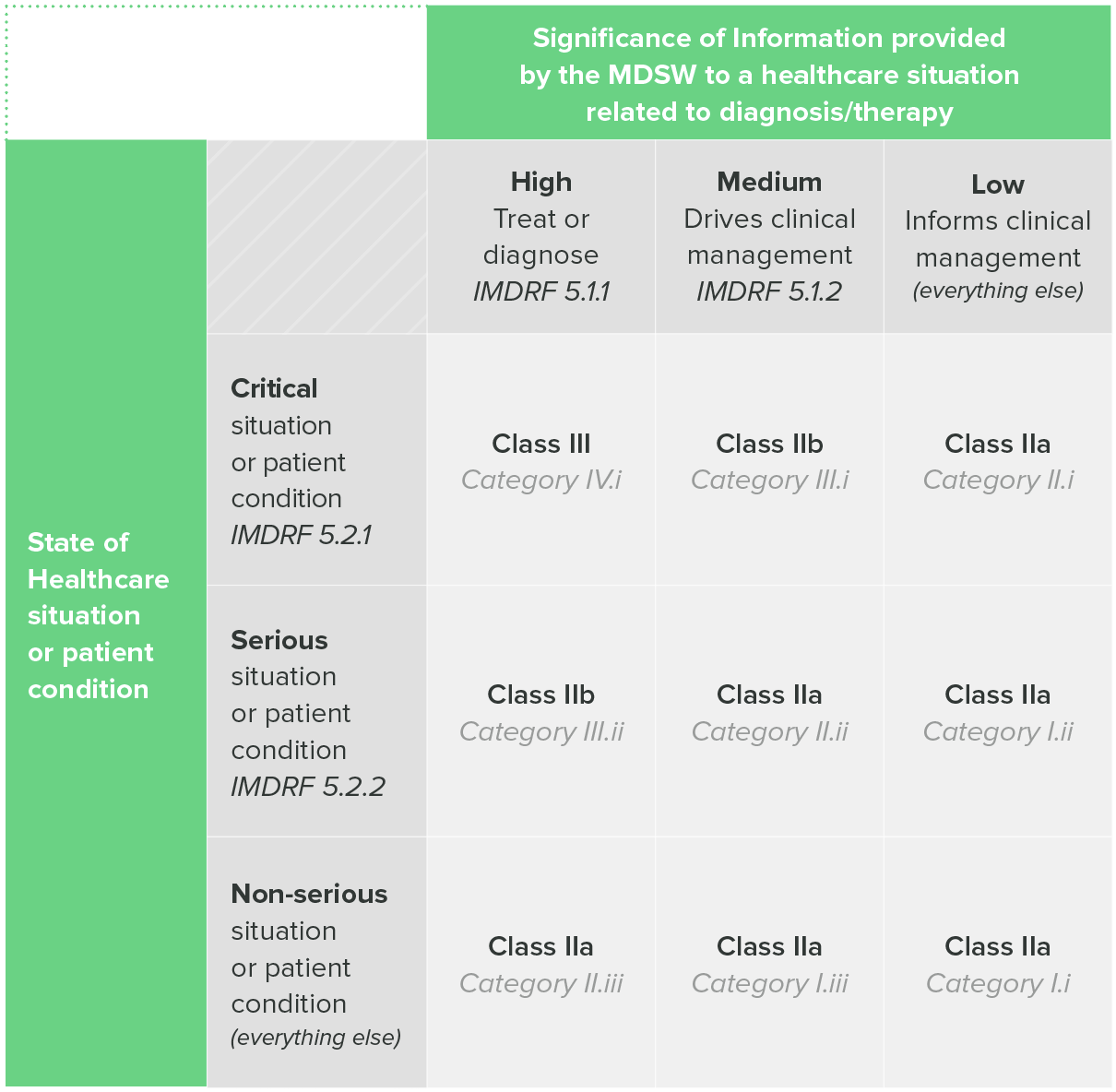
If you are navigating Rule 11 for the first time, working through the IMDRF framework first can help you think through your classification before applying EU-specific criteria.
IEC 62304 software safety classification
Separate from any regulatory risk class is the software safety classification established by IEC 62304, the international standard for medical device software lifecycle processes. IEC 62304 uses three safety classes based on the severity of injury a software failure could cause:
-
Class A: No injury or damage to health is possible.
-
Class B: Injury is possible, but not serious.
-
Class C: Death or serious injury is possible.
It's important to understand both what this classification is and what it isn't. Software safety classification under IEC 62304 is not a determination of how safe your software is. It is the basis for determining how rigorous your development process must be. The actual safety of your device depends on your design and development processes, not on a safety classification.
And keep in mind, IEC 62304 safety class does not directly correspond to FDA or EU risk class. There is a strong correlation, but the two systems are not equivalent. It's likely that Class C software would also be a Class III device in the US or the EU, but it's possible that you could have a software product that carries a Class C safety classification yet falls into a lower regulatory risk class. The two determinations are made independently, using different criteria.
However, IEC 62304 is an FDA-recognized consensus standard and use of the standard is considered best practice for meeting software safety requirements.
How the classification systems relate to each other
One of the most common mistakes SaMD manufacturers make is assuming that any classification maps neatly onto another. They do not. You should never use your IEC 62304 safety class as a proxy for your FDA or EU risk class, or vice versa.
What the systems share is a common underlying concern for the potential for harm to patients and users. A product with a high potential for harm will generally carry high classifications across all systems. But the specific thresholds, criteria, and implications differ, and each system must be worked through on its own terms.
The practical advice here is to treat each classification as a separate analysis. Work through the FDA system for your US submission, the EU MDR system for your EU submission, and IEC 62304 safety classification as part of your software development process. Use the IMDRF framework where it helps you think through EU classification. And document each determination carefully, because regulators will want to see your reasoning.
While it may sound daunting, this can actually benefit you by impacting your regulatory strategy. It may be easier to go to market in some regions than in others. Some regulatory frameworks may even treat the same software as a completely different device or not as a device at all (remember the CDS criteria in the US). If you figure out your different classifications and plan ahead, it may actually change your overall strategy.
BONUS RESOURCE: Click here to download your free Cybersecurity Gap Assessment Checklist!
How Greenlight Guru supports SaMD classification and compliance
Classification is just the beginning. Once you know where your software falls, you need an eQMS capable of meeting the obligations that come with it and keeping pace as your product evolves.
Greenlight Guru is built specifically for medical device teams, with purpose-built tools for managing design controls, risk, and software release in a single connected system. That means your classification decisions, risk analysis, and development documentation are all traceable to one another, which is exactly what regulators expect to see. Whether you're working toward FDA clearance, CE marking, or both, Greenlight Guru helps you build the evidence you need without building it from scratch in spreadsheets.
To learn more about how Greenlight Guru supports SaMD and SiMD teams, get your free demo today!
Cybersecurity Gap Assessment Checklist

.png?width=575&name=Quality%20Lead%20Magnet%20Slide-in%20(5).png)
.png?width=250&height=321&name=Quality%20Lead%20Magnet%20Slide-in%20(5).png)



