








The global medical device industry is a space where forward thinking meets problem solving, all for the same purpose: to improve the quality of life. However, if you’re going to compete on a global scale, you need to embrace the concept of “Think Globally, Act Locally,” especially when it comes to regulatory compliance with the IVDR.
In this ultimate guide, we’ll break down IVDR, the reasons behind the new European regulations, the numerous annexes and guidelines associated with IVDR, and paint the full picture that shows you how you can transition to IVDR in the EU market.
How does IVDR differ from IVDD?
What are the key components of IVDR?
Notified bodies & CE marking
Economic operators
Technical documentation
Performance evaluations, performance studies, and post-market performance follow-up
Post-market surveillance
Device labeling and UDI
EUDAMED
Quality management system requirements
How are devices categorized under IVDR?
How to classify your IVD device
IVDR compliance is made easy with Greenlight Guru
FREE EBOOK: Download your PDF copy of the Ultimate Guide to IVDR by clicking here.
What is IVDR?

The IVDR or In Vitro Diagnostic Regulation (IVDR 2017/746) is the European Commission’s legislation for in vitro diagnostic (IVDs) medical devices. It consists of a set of regulations that govern the manufacturing, distribution, and use of in vitro diagnostic medical devices in the European Union.
Published on May 25, 2017, IVDR introduces an expansive set of new rules for how in vitro diagnostic devices are regulated in the European Union (EU).
The goal was to replace decades-old legislation with regulations that are more in line with the latest technological advances and medical science, as well as create a more transparent regulatory framework that helps improve patient safety.
What are IVDs?

In vitro diagnostics are clinical tests that analyze biological samples, such as blood, fluid, or tissue. IVDs are a common tool in healthcare, used in detecting disease or infection, measuring the concentration of specific analytes, or monitoring individual patient health.
In vitro means the tests are performed outside the body using test tubes, test kits, or related lab equipment. In fact, IVD samples can be collected and analyzed both in traditional healthcare settings, such as hospitals and testing labs, as well as remotely, such as at home or in the field.
Thanks to the versatility of in vitro tests, IVDs have played a huge role in the rise of telemedicine and remote patient monitoring. During the COVID-19 pandemic, IVD products made it possible for millions of people to test themselves for the virus from the safety of their own home. IVD products have also become a major resource for providing treatment to underserved, low-income populations.
Some other examples of IVDs are:
-
HIV testing
-
COVID tests
-
Pregnancy tests
-
Blood glucose monitors
-
Cancer diagnostics
-
Clinical chemistry assays
-
Companion diagnostics
-
Devices for blood grouping
-
Devices for the detection of infectious agents
-
Devices for human genetic testing
-
Devices for tissue typing
-
Immunoassays
IVDR defines an in-vitro diagnostic medical device as,
any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body solely or principally for the purpose of providing information on one or more of the following:
(a) concerning a physiological or pathological process or state;
(b) concerning congenital physical or mental impairments;
(c) concerning the predisposition to a medical condition or a disease;
(d) to determine the safety and compatibility with potential recipients;
(e) to predict treatment response or reactions;
(f) to define or monitor therapeutic measures.
What is IVDR replacing?

IVDR was created as a replacement for the EC’s previous legislation, the In Vitro Diagnostic Medical Device Directive (IVDD). Passed in 1998, IVDD was groundbreaking in setting requirements for performance, safety, and quality of IVD products in the EU. However, experts worried it lacked the scope needed to hold manufacturers accountable for patient safety.
Not only that, but there was also a growing concern that the Directive could quickly become outdated. The 2000s were a time of rapid advances in MedTech and any regulations would need to stay fresh to keep in pace with the industry, and in vitro diagnostics were certainly no different. As the years passed, the experts saw just how accurate their predictions had been.
In 2010, while the IVDD was awaiting review, the PIP scandal took place in the medical device sector. This was the tragic series of injuries and deaths in which a manufacturer deliberately used industrial grade silicone to make breast implants. While it was technically in violation of the then-current framework for device safety, none of the checkpoints in place were enough to prevent such a crime against the public’s trust.
The impact of this scandal and the attempts to remedy it can clearly be seen in the text of the IVDR. Both the IVDR and the EU Medical Device Regulation (EU MDR) aim to address concerns around patient safety and, as a result, the text includes increased expectations for clinical evidence, scrutiny of manufacturers data, greater expectations for both competent authorities and notified bodies, traceability throughout the supply chain, and greater transparency.
How does IVDR differ from IVDD?

IVDR provides stricter oversight in many areas as compared to the EU’s previous IVDD. That said, there are a considerable number of requirements copied over from the Directive, many of which function as expansions on preexisting IVDD Articles.
Some of the IVDD elements that have remained unchanged in the new IVDR are:
-
Essential requirements
-
Technical documentation
-
Conformity assessment
-
Choice between assessment levels of quality management system
-
Registration requirements for manufacturers
-
Vigilance
Perhaps the most notable change is in who this legislation applies to. Under IVDD, only about 10-20% of IVD manufacturers needed a notified body's statement of conformity, and most were allowed to self-certify.
Under IVDR, though, that number jumps to 80-90% of IVD products expected to fall within one of the risk classifications which requires the involvement of notified bodies. That, coupled with an expanded definition of in vitro diagnostic devices that includes genetic testing and related software, is already slowing down the approval process.
However, this is all part of an overarching shift towards a greater emphasis on a complete product lifecycle rather than just the pre-approval steps. We can see that in a few different forms, such as the increased requirements for post-market surveillance, the greater supervision for notified bodies, and entirely new employee and stakeholder roles.
What are the key components of IVDR?

IVDR is an extensive and exhaustive list of standards, annexes, and guidance that manufacturers are expected to know all the ins and outs of. Even today, more than five years after its publication, IVDR continues to be a source of confusion and frustration for manufacturers.
With that in mind, we’ve broken down the key components of IVDR you need to know, and provided additional resources for areas that deserve a double click.
Notified bodies & CE marking
In the EU, all medical devices must receive a CE marking before being sold or marketed in the member states. CE markings are indicators which communicate that the product meets the region’s requirements for health, safety, and environmental protection.
The only way to receive a CE marking for IVDR products will require the involvement of a notified body, who will:
-
Audit the manufacturer’s quality management system (QMS)
-
Review the technical documentation for the different classes of devices
-
Issue CE certificates
Remember that the goal with IVDR is to ensure transparency and accountability, as well as ensure no single poor performance point in the product lifecycle undoes the commitment to mitigating risk at all turns.
A complete listing of the notified bodies that are officially designated is maintained on the NANDO (New Approach Notified and Designated Organisations) database.
And because it’s a full product lifecycle, the job is not done there for notified bodies, as they will take on more responsibility in enforcing regulations through annual on-site assessments as well as unannounced audits of manufacturing processes, subcontractors, and suppliers.
Economic operators
Another layer of accountability in IVDR can be found in the new defined roles for economic operators. Think of this as a full product lifecycle approach across the whole supply chain, with considerations and requirements made for anyone who is involved in the design, manufacturing, or distribution of IVD products.
IVDR defines several of these economic operator roles directly in text:
‘Manufacturer’ means a natural or legal person who manufactures or fully refurbishes a device or has a device designed, manufactured or fully refurbished, and markets that device under its name or trademark.
‘Importer’ means any natural or legal person established within the Union that places a device from a third country on the Union market.
‘Distributor’ means any natural or legal person in the supply chain, other than the manufacturer or the importer, that makes a device available on the market, up until the point of putting it into service.
‘Authorised representative’ means any natural or legal person established within the Union who has received and accepted a written mandate from a manufacturer, located outside the Union, to act on the manufacturer’s behalf in relation to specified tasks with regard to the latter’s obligations under this Regulation.
The IVD regulation also establishes that manufacturers must appoint an employee to be their Person with Regulatory Responsibility (PRRC).
The duties for the PRRC are outlined in Article 15(3), stating that this individual is responsible for ensuring that:
(a) the conformity of the devices is appropriately checked, in accordance with the quality management system under which the devices are manufactured, before a device is released;
(b) the technical documentation and the EU declaration of conformity are drawn up and kept up-to-date;
(c) the post-market surveillance obligations are complied with in accordance with Article 10(9);
(d) the reporting obligations referred to in Articles 82 to 86 are fulfilled; NOTE: This refers to Vigilance reporting, i.e. serious incident, field safety corrective action, and incident trend reporting);
(e) in the case of devices for performance studies intended to be used in the context of interventional clinical performance studies or other performance studies involving risks for the subjects, the statement referred to in Section 4.1 of Chapter II of Annex XV is issued.
Technical documentation
Annexes II and III provide specific requirements for technical documentation, specifying that the technical file “shall be presented in a clear, organized, readily searchable, and unambiguous manner.”
Let’s take a look at what documentation is required in a technical file:
-
Device description and specifications
-
Reference to previous and similar generations of the device
-
Package labeling and instructions for use (in appropriate languages)
-
Product design and manufacturing information (including listing of all supplier and contract manufacturer sites)
-
General safety and performance requirements (Annex I)
-
Benefit-risk analysis and risk management plan
-
Product verification and validation, including:
-
Specimen type/handling
-
Analytical performance
-
Interfering endogenous/exogenous substances investigated
-
Clinical performance
-
Performance of self-testing devices/near-patient testing devices
-
Scientific validity
-
-
Pre-clinical and clinical data (performance evaluation report)
-
Post-market surveillance plan and reports
Ensuring your technical documentation is fully prepared is crucial for audits by your notified body as part of the CE marking process. To learn more check out this deeper dive into maintaining your technical file the right way.
Performance evaluations, performance studies, and post-market performance follow-up
One of IVDR’s most notable changes is in its requirements for clinical data. Manufacturers are responsible for providing this data for the purpose of safety and performance claims throughout the product lifecycle. Manufacturers should expect a greater level of scrutiny, and plan to implement methods to proactively collect and evaluate data.
IVD manufacturers should collect, analyze, and provide data using the following tools:
-
Performance Evaluations (PE) — Performance evaluations must be thorough and take into account both favorable and unfavorable data. These should be conducted and documented in accordance with Article 56 and Annex XIII (Part A).
-
Performance Evaluation Plan (PEP) — Another notable aspect of IVDR is the expectation for early planning in performance evaluations. To stay in-step with these requirements, manufacturers should proactively establish a performance evaluation plan before gathering any and all clinical data.
-
Clinical Data Sources — With a PEP in place, manufacturers will need to identify any and all sources for its clinical data, specifically those relating to the safety, performance, and intended purpose of an IVD device.
-
Clinical Evaluation Assessment Report (CEAR) — Manufacturers must ensure that the clinical evaluation assessment report is built into their quality management process. Additionally, CEARs must be connected to and aligned with any other components of technical documentation.
-
Clinical Performance Studies — In the event that elements of an IVD device’s performance cannot be verified through analytical performance studies, related device literature, or routine diagnostic testing, IVDR will require manufacturers to conduct clinical performance studies.
-
Performance Evaluation Report (PER) — PERs are needed to document data related to scientific validity, clinical performance, and analytical performance. Along with any CEARs, performance evaluation reports must be included with the same technical documentation that is used for the conformity assessment.
-
Post-market Performance Follow-up (PMPF) — Keeping in-line with IVDRs focus on total product lifecycle, manufacturers must confirm the safety and performance and continued acceptability of product risks with a post-market performance follow-up.
Post-market surveillance
IVDR requires manufacturers to dedicate a high level of attention to monitoring a device’s performance once it’s on the EU market. Doing so will require a post-market surveillance (PMS) plan.
Annex III 1(b) provides manufacturers a list of minimum required categories to be included in a PMS plan:
-
Indicators and threshold values for continuous reassessment of the benefit-risk analysis and risk management (Section 3 of Annex I);
-
Methods and tools to investigate complaints and analyze market-related experience collected in the field;
-
Methods and protocols to manage the events subject to the trend report as provided for in Article 83, including the methods and protocols to be used to establish any statistically significant increase in the frequency or severity of incidents as well as the observation period;
-
Methods and protocols to communicate effectively with competent authorities, notified bodies, economic operators and users; —
-
PMS procedures to fulfill the manufacturers obligations laid down in Articles 78, 79 and 81; — systematic procedures to identify and initiate appropriate measures including corrective actions; —
-
Trace and identify devices for which corrective actions might be necessary; and — a PMPF plan as referred to in Part B of Annex XIII, or a justification as to why a PMPF is not applicable.
For Class A and B devices, IVDR will require a postmarket surveillance report, which includes a summary of results and conclusions about post-market surveillance data defined in the PMS plan, as well as the rationale and description of any corrective or preventive actions taken.
For Class C and D devices, you’ll need to perform a Periodic Safety Update Report, which builds upon the data collected for the PMSR and additionally includes key findings from the PMPF, conclusion of the benefit/risk determination, and data on sales figures, user populations, and frequency of use.
Device labeling and UDI
The new IVDR has expanded labeling requirements to increase transparency and traceability of devices.
IVD device labeling must include:
-
Device name
-
Trade name
-
Standardized symbol affirming status as in vitro diagnostic medical device
-
Statement of Intended purpose
-
Contact information of authorized EU representative (for non-EU based manufacturers)
-
Time limit for safe device use (i.e., year and month)
-
All user warnings
-
Serial number
-
Lot number
-
Type of specimen(s) required to perform the test (e.g., blood, urine, saliva)
-
Links to electronic instructions for use (eIFUs) and company website
-
Indication of any special storage and/or handling conditions
-
Indication of sterile state of the device or any special microbial state
-
Indication if the device is intended for single use
-
Indication if device is intended for self-testing, or near-patient testing
IVDR also requires the integration of the unique device identifier (UDI). UDI allows for the unambiguous identification of specific devices on the EU market and enables better traceability and monitoring by competent authorities. In addition to being added to device labeling, the UDI information must also be listed on the declaration of conformity and uploaded to the European database for medical devices (EUDAMED).
EUDAMED
As part of the new IVDR, the European Commission developed a secure database called EUDAMED to improve transparency and coordination of information regarding medical devices on the EU market.
The EUDAMED system is comprised of six modules:
-
Actor Registration: Enables economic operators (i.e., manufacturers, authorized representatives, and importers) to register their information
-
Unique Device Identification (UDI): Maintains device-specific information. Economic operators are responsible for managing all UDI attributes and transferring the data to EUDAMED
-
Certificates: Maintains EU certificates for each product group
-
Clinical Investigation: Maintains information regarding the collection and analysis of clinical data
-
Vigilance: Maintains serious incident reports
-
Market Surveillance: Maintains post-market surveillance reports
EUDAMED provides transparency, particularly when it comes to serious incident and corrective action reporting. Under IVDR, manufacturers will need to report all serious incidents and field safety corrective actions to competent authorities via the EUDAMED database.
Once the manufacturer becomes aware of the incident, these serious incidents must be reported within these timeframes:
-
If it represents a serious threat to public health, incident must be reported immediately, but no later than 2 days post incident
-
If it involves a death, incident must be reported Immediately, but no later than 10 days post incident
-
If it has little to no consequence to the patient or user, the incident must be reported immediately, but no later than 15 days post incident
Quality management system requirements
One of the best offenses is a good defense, which is exactly why focusing on your quality management system (QMS) is imperative to IVDR navigation. Leaning on the established processes for design controls, risk management, design transfer, and manufacturing best practices can smooth the transition to an IVDR-compliant QMS.
IVDR requires a QMS to address the following:
-
Defined strategy for regulatory compliance, including compliance with conformity assessment procedures and procedures for the management of device modifications
-
Designated PRRC
-
System for identifying all applicable general safety and performance requirements
-
Product realization—planning, design, development, production, and service provision
-
Management of resources (i.e., infrastructure and equipment) and supply chain
-
Development of a risk management plan
-
Process for post-market surveillance
-
Process for performance evaluation and post-market performance follow-up
-
Periodic safety update reporting
-
System for serious incidents and corrective action reporting
-
Processes and system to manage corrective and preventive actions (CAPAs)
-
Verification process for CAPA system effectiveness
-
Processes and procedures to fulfill UDI requirements
-
Processes for product improvement, monitoring and measurement of output, and data analysis
-
Processes for managing and ensuring traceability of communications with competent authorities, notified bodies, economic operators, customers, and other stakeholders
How are devices categorized under IVDR?

Although in vitro diagnostics are non-invasive, they are still categorized as a medical device based on their intended use in informing healthcare decisions. This introduces a significant level of risk, as inaccurate IVD results can lead to medical errors or even death. As such, in vitro diagnostic devices are regulated in medical device markets across the globe.
Under IVDR, in vitro diagnostic devices are categorized by risk, much the same way that other medical devices are grouped under EU MDR. However, IVDs also are used with biological material that has been removed from the body to test the transmissible agents within the biological material. With this in mind, IVDs are also categorized based on the potential risk they may pose to public health.
IVDR establishes four risk classes based on both patient and public health risk:
-
Class A - Low patient and public health risk. This includes specimen receptacles, laboratory instruments, and buffer solutions.
-
Class B - Moderate patient risk and/or low public health risk. Class B devices include IVDs for self-testing such as pregnancy tests, fertility tests, and cholesterol tests. Class B is also the default classification for IVDs that are not covered by any other rules.
-
Class C - High patient risk and/or moderate public health risk. Class C devices include IVDs that are intended to be used for detecting an infectious agent without a high risk of propagation, or for detecting the presence of an infectious agent with the potential to cause death or severe disability in the case of an erroneous result.
-
Class D - High patient risk and high public health risk. This device class includes IVDs that detect or are exposed to life-threatening transmissible agents or transmissible agents and infectious diseases with a high risk of propagation.
It’s important to note that manufacturers must meet the applicable underlying safety and performance requirements set out in Annex I of the IVDR, regardless of the product's classification.
That said, an IVD device’s classification establishes whether or not certification by a notified body is required. The classification also determines which conformity assessment route — Annex IX, X, or XI — manufacturers will need to take. Each one of these has different requirements for product verification. Annex IX requires a review of QMS and Technical Documentation; Annex X requires a Type Examination; and Annex XI calls for a Product Conformity Verification.

How to classify your IVD device
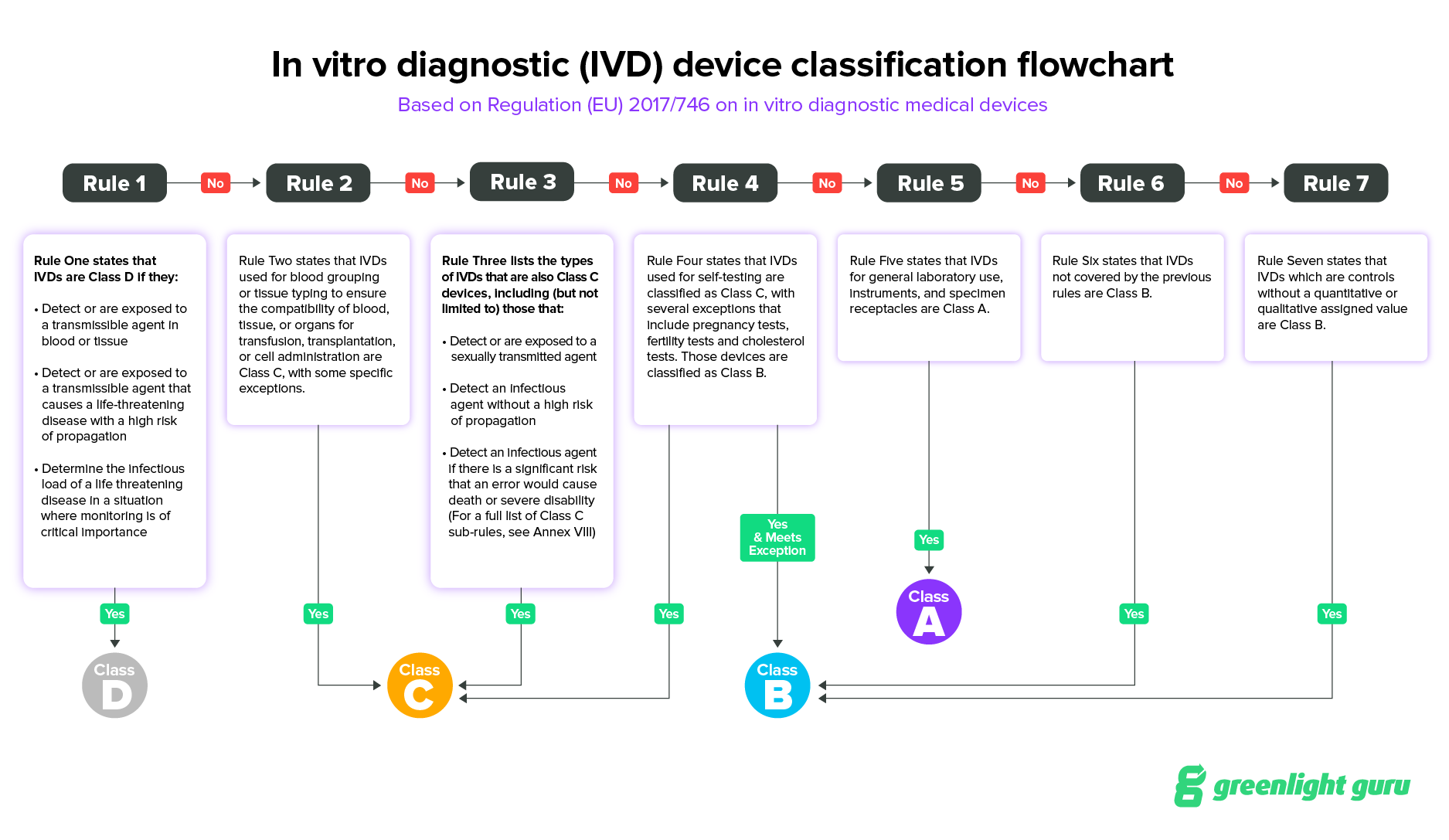
IVDR outlines seven rules for classification and provides guidance in Annex VIII for IVD manufacturers to identify the risk class of IVD devices.
Rule One states that IVDs are Class D if they:
-
Detect or are exposed to a transmissible agent in blood or tissue
-
Detect or are exposed to a transmissible agent that causes a life-threatening disease with a high risk of propagation
-
Determine the infectious load of a life threatening disease in a situation where monitoring is of critical importance
Rule Two states that IVDs used for blood grouping or tissue typing to ensure the compatibility of blood, tissue, or organs for transfusion, transplantation, or cell administration are Class C, with some specific exceptions outlined that will move the IVD into Class D..
Rule Three lists the types of IVDs that are also Class C devices, including (but not limited to) those that:
- Detect or are exposed to a sexually transmitted agent
- Detect an infectious agent without a high risk of propagation
- Detect an infectious agent if there is a significant risk that an error would cause death or severe disability (For a full list of Class C sub-rules, see Annex VIII)
Rule Four states that IVDs used for self-testing are classified as Class C, with several exceptions that include pregnancy tests, fertility tests and cholesterol tests. Those devices are classified as Class B.
Rule Five states that IVDs for general laboratory use, instruments, and specimen receptacles are Class A.
Rule Six states that IVDs not covered by the previous rules are Class B.
Rule Seven states that IVDs which are controls without a quantitative or qualitative assigned value are Class B.
IVDR implementation plan

Whether your IVD device is new to the EU market, or you’re an established company looking to transition to IVDR compliance, there are a few tasks that should be your top priorities:
-
Contract with a notified body immediately. In 2021, the EC warned about a “grave shortage of notified body capacity.” At the time of writing there are currently only seven notified bodies listed on NANDO, resulting in significant delays, so the sooner you can get your product onto their radar the better.
-
Review GSPR guidelines and classifications. Before you head down a time-consuming path, it’s a good idea to make certain your IVD device is adhering to the right standards, and that you are following the applicable regulatory requirements for your IVD classification.
-
Break down team silos within your organization. IVDR compliance will impact nearly every area of your business, and documentation needs to be completed for just about every part of the CE marking process, so traceability and communication among teams is imperative.
-
Perform gap analysis on all departments and activities. How will IVDR change your SOPs, QMS, or go-to-market strategies? The only way to be sure is by performing a gap analysis that allows you to see the big picture, prioritize areas that need the most work, and accurately set budgetary expectations. You can also use the our IVDR gap analysis tool, which provides an easy method to understand, assess and execute the necessary changes for compliance.
-
Plan (and budget) for delays. As mentioned earlier, timelines for implementation have changed significantly since 2017. While you want to push ahead, there may be some wait time before you’re able to market your device, and your stakeholders will want to be prepared for those costs.
-
Work your supply chain magic and manage it. Under the IVDR framework, you are responsible as an economic operator for not only your compliance, but the compliance of your suppliers. Once you have established an Approved Supplier List (ASL) make sure to ask if they are certified with the relevant technical standards. If not, you’ll need to incorporate training and internal audits to make sure you’re not getting held up by something out of your hand.
FREE EBOOK: Download your PDF copy of the Ultimate Guide to IVDR by clicking here.
IVDR compliance is made easy with Greenlight Guru

The move to a risk-based classification system under IVDR means that risk management is more important now than ever for IVD manufacturers.
At Greenlight Guru, we understand the critical role that risk plays in classifying and marketing a safe and effective device.
Greenlight Guru is the only out-of-the-box QMS solution designed to help IVD companies navigate the medical device regulatory environment and get products to market faster, with less risk.
From reagents to instruments to systems intended for use, you can document your design controls, build your hierarchies of components and subcomponents, and manage your product families.
Greenlight Guru customers often speak about their newfound ability to simplify regulatory compliance and provide a single source of truth by connecting the management of all product development, regulatory, and quality processes from documentation and design controls to submission and ongoing compliance.
Get your free demo of Greenlight Guru’s Quality Management Software to see just how easy we make risk analysis and IVDR classification for your device.
Another way Greenlight Guru supports you in bringing safe medical devices to market, is with our e-clinical solution: Greenlight Guru Clinical, the ideal toolbox for post-market activities.
Greenlight Guru Clinical is our Electronic Data Capture (EDC) Software designed specifically for the MedTech industry. With our EDC software, you will be empowered to collect high quality data for performance evaluations (PE) or PMPF while at the same time complying with regulatory requirements.
Get your free demo of Greenlight Guru Clinical, and see for yourself how it can streamline your clinical data collection and management for safety and performance studies.
About the Co-authors

Laura Maher, Medical Device Guru, Greenlight Guru
Laura Maher is a Medical Device Guru who has spent her career working in a variety of different roles ranging from product development, process improvement, and quality system management.
Laura learned early in her career the importance of product quality and the impact it has for patients.
She loves to help customers find success in bringing safe, effective, and innovative medical devices to market.
 Niki Price, Medical Device Guru, Greenlight Guru
Niki Price, Medical Device Guru, Greenlight Guru
Niki Price is a Medical Device Guru who has spent her entire career working with different types of medical devices.
She began her journey in production, which is where she discovered how important and fulfilling this line of work was to her!
Spending time in both Quality and R&D, she enjoys the product development process and witnessing great results as a product of great design. Niki hopes to continue to her journey by helping others build quality into their products and processes.
The Ultimate Guide To IVDR


.png?width=250&height=324&name=Ultimate%20Guide%20to%20IVDR%20(cover).png)



