
Table of Contents
PART I: CASE FOR QUALITY VOLUNTARY PILOT PROGRAM
VISION BEHIND THE CASE FOR QUALITY PROGRAM
INITIATION AND DEVELOPMENT OF CASE FOR QUALITY
CASE FOR QUALITY PILOT PROGRAM OVERVIEW
VALUE DERIVED ACROSS INDUSTRY STAKEHOLDERS
WHAT IS INVOLVED IN THE VOLUNTARY PILOT PROGRAM?
KEY OBJECTIVES OF CASE FOR QUALITY
PART II: WHY THE CASE FOR QUALITY PROGRAM MATTERS
SHIFTING FROM COMPLIANCE TO OPERATIONAL EXCELLENCE
CREATING A VIRTUOUS CYCLE OF IMPROVEMENT
THE CAPABILITY MATURITY MODEL INTEGRATION
IMPLEMENTING THE MATURITY APPRAISAL MODEL
CASE FOR QUALITY PROGRAM PROCESS
FDA INSPECTION VS. CMMI APPRAISAL
COMPLIANCE INDICATORS VS. QUALITY INDICATORS
PART III: COMPLIANCE VS. QUALITY METRICS
COMPLIANCE VS. QUALITY INDICATORS
ENHANCING VISIBILITY AND INNOVATION
APPLYING PROVEN METHODS IN FUTURE PROGRAMS
PART IV: HOW THE PILOT ADDRESSES VALIDATION PROCESSES
BREAKING DOWN INDUSTRY BARRIERS
FDA’S POSITION ON AUTOMATION TOOLS
STREAMLINING VALIDATION PROCESSES
IMPACT ON QUALITY ASSURANCE LANDSCAPE
EMBRACING AUTOMATED COMPUTER SYSTEM VALIDATION TOOLS
THE FUTURE OF CASE FOR QUALITY
Foreword
The Medical Device Innovation Consortium (MDIC) has had the privilege of partnering with the FDA Center for Devices and Radiological Health (CDRH) to advance the Case for Quality, a transformational initiative to shift the medical device industry from a focus on regulatory compliance to a focus on quality maturity.
This kind of cultural shift doesn’t happen overnight. MDIC, FDA and our industry partners have worked together to develop tools and methods to encourage and appropriately incentivize quality practices.
Beyond the tools, MDIC has sought to cultivate trust between medical device manufacturers and the FDA. That trust is fundamental to developing a culture based on a mutual commitment to quality maturity practices, rather than “check the box” compliance activities.
In December 2017, CDRH launched the Voluntary Medical Device Manufacturing and Product Quality Program Pilot, utilizing a maturity model refined in collaboration with the Capability Maturity Model Integration (CMMI) Institute, MDIC, and regulatory and industry partners. The maturity model is leveraged as a resource for medical device organizations to measure their capability to produce high quality, safe and effective devices.
This measurement can then be used by organizations to drive targeted continuous improvement activities throughout their facilities. For manufacturers who complete the independent (third party) appraisal of quality maturity, the FDA will adjust their engagement activities and modify their submission requirements and routine inspection plans.
Industry participation is critical to long-term implementation of the maturity model as an alternative to the traditional path of a routine FDA inspection. Participation in the pilot requires an investment, both of personnel and money.
However, companies will receive many valuable benefits. Participating companies can expect to improve organizational processes and reduce variability that could lead to reduced costs of quality, decreased rework, and increased return on investment.
The FDA will also benefit from this program by potentially reducing the internal resources required for evaluation of inspections and manufacturing review submissions. The combined focus on safety and quality can be a win-win for both FDA and manufactures as we advance the health and safety of patients.
MEDICAL DEVICE INNOVATION CONSORTIUM (MDIC)
Preface
Greenlight Guru launched an exclusive webinar series alongside FDA’s center for medical devices (CDRH) to bring industry awareness to their Case for Quality program.
In 2011, FDA’s Center for Devices and Radiological Health (CDRH) launched its Case for Quality initiative to transform its focus from primarily regulatory compliance for medical device manufacturing to a focus on quality, inclusive of regulatory compliance.
An in-depth review of device quality data and feedback from both the FDA and industry stakeholders revealed that FDA’s compliance requirements did not ensure uniformity in device quality across the ecosystem.
For instance, a device could be regulatory compliant, but also low quality; conversely, it could be high quality, yet non-compliant and therefore non-marketable. It became clear to FDA that compliance was not enough to ensure quality.
Since this discovery, FDA has established mechanisms for stakeholder engagement, developed the Voluntary Manufacturing and Product Quality program, and is piloting a streamlined 510(k) submission review to support this notion of continuous improvement.
The Case for Quality program has allowed FDA to work closely with medical device stakeholders — manufacturers, healthcare providers, patients, payers and investors — to shift the agency’s focus from serving as a regulatory-only role to also becoming as a collaborative industry partner.
The driving force behind the Case for Quality initiative lies in its focus on continuous improvement for medical device companies. With the support and advocacy of FDA, industry stakeholders will be enabled to develop high quality products and ultimately benefit the patients who use them.
This comprehensive eBook provides an overview of the Case for Quality initiative and the multi-part webinar series Greenlight Guru hosted in partnership with FDA.
I encourage you to learn more about this program because, more than anything else, embracing these approaches and methodologies will have a profound impact on your products and processes. Above all else, patients receiving your devices will impact from this True Quality approach.
JON SPEER
Part I: Case For Quality Voluntary Pilot Program
Throughout most of my 20+ years in the medical device industry, I’ve had frequent first-hand experience with the contentious relationship between FDA and medical device companies. And historically, the nature of these interactions have over-emphasized the importance of being compliant to the regulations.
Yes, of course, compliance is important. But at what cost does it come with to the product and process quality?
Often times, we as medical device professionals make choices about what to do and how to do it – more in the spirit of satisfying the needs of regulatory bodies, rather than what makes sense for your business. In doing so, have product and process qualities suffered? Sadly, probably so.
Striving to be compliant with the rules and regulations have influenced and dictated behaviors and thwarted the embrace of best practices. To its credit, FDA CDRH observed this finding as a significant issue, and subsequently initiated the Case for Quality program.
At Greenlight Guru, once we had a better understanding of the program, we were determined to team up with MDIC and FDA to help spread the word.
Through a multi-part webinar series with Cisco Vicenty, FDA CDRH Case for Quality Program Manager, we’ve been able to share an overview of the Case for Quality program and host status update webinars.
For the first installment of our four-part series, I spoke with FDA’s Case for Quality Program Manager, Francisco “Cisco” Vicenty, to learn more about at the what, why and how behind this change in regulatory paradigm.

Vision Behind The Case For Quality Program
The Case for Quality program springs from CDRH’s overarching vision of U.S. patients having access to high-quality, safe, and effective medical technologies.

Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
This vision is the driving force behind how FDA has shifted its focus and the way it now approaches regulatory programs.
The bottom line is that it’s all about the patient. As an industry, we need to work together to drive the best possible public health outcomes.
For FDA, this has meant a systemic shift in focus to establish the means necessary to achieve those aforementioned outcomes. Rather than focusing on regulatory as a sort of “checkbox” activity, a shared focus on quality is just as important.
The collaboration and engagement of all stakeholders is necessary to shift the focus from compliance to quality. It’s not enough to say that either the agency or manufacturers are to blame for a delay in innovative solutions, a whole community needs to be brought into the fold.
Innovation is key, particularly being able to innovate quickly, with a collective goal for continuous improvement. The regulatory framework needs to be adaptive and responsive in order to enable optimal results for innovation and improvement.
Our Case for Quality webinar series presenter, Cisco, highlighted some key statistics that help to illuminate why this initiative is so important. After analyzing the metrics on the program participants, the numbers yield a realistic goal well within the reach of the agency to enable this new level of quality and innovation.

Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
Initiation and Development of Case For Quality
Year after year, the FDA would go out and perform inspections of medical device companies. In doing so, the agency found itself issuing a staggering number of form 483 observations to companies for non-compliances. This was especially concerning to FDA since the same compliance issues kept recurring, with no real change or improvements being made to resolve the problem.

From 2010-2011, FDA performed a full analysis, finding several factors at hand which they believed were driving these noncompliance issues. As a result, there became a predominant focus on compliance itself, with three specific factors:
- Industry focus was hinged on solely meeting regulatory requirements (think “compliance”), rather than adopting the best quality practices.
- There was overall low investment in automation and digital technologies, which is known to curtail poor manufacturing processes and lead to more responsive learning and action from entities that utilize them.
- There was no competitive market around medical device quality.
To support the shift from compliance-focused to also include a quality-focused mindset, CDRH awarded the Medical Device Innovation Consortium (MDIC) a contract in 2015. This partnership would allow MDIC to pool people, resources, and ideas to develop and research the tools, methods, and practices that would make it possible to assess this new medical device quality initiative.
With the support its partnership with MDIC, FDA and the Case for Quality program is able to offer a unique forum for medical device stakeholders to move beyond baseline regulatory compliance activities, in order to collaboratively develop sustained predictive practices to advance medical device quality and safety for better patient outcomes.

This realization resulted in a collaborative undertaking by FDA and MDIC to create this new industry shift that would focus on organizational excellence.
FDA also concluded that it would need to be able to adjust its own agility, responsiveness, and adaptability for this new environment for quality to not only work, but also yield the intended outcomes.

This could be done through several different ways, such as simplifying and error-proofing processes, as well as allowing for continuous, rapid improvement. The assessment of organizational performance needed to shift from an exclusively investigative role in order to build the trust with organizations and drive meaningful connections within quality systems.
Any preceding processes that could be considered overly burdensome would need to be evaluated for simplification in order to enable quality outcomes that emphasize operational excellence.
Case For Quality Pilot Program Overview
In December of 2017, CDRH launched the Voluntary Medical Device Manufacturing and Product Quality Pilot Program utilizing a maturity model refined in collaboration with the Capability Maturity Model Integration (CMMI) Institute, MDIC, and other regulatory and industry partners.
This program enables medical device stakeholders to collaborate on the enhancement of medical device quality and patient safety. With a proven, third-party maturity appraisal methodology, the pilot program has a number of participants that opted to sign up. The initial pilot started January 2, 2018 and concluded on December 28, 2018.
The methodology used for the participation from medical device companies included a capability maturity model integration framework to assess a company’s ability to produce high-quality devices that adhere to the highest level of safety for its end users.
In order to be eligible to participate in the pilot, companies had to provide a record of compliance. Serving as the federal regulatory agency for the U.S., FDA has a duty to protect its citizens, so enforcing compliance will always be a priority function of that role.
With that said, this pilot program offers some appealing perks to its participating medical device companies. FDA set up the program to let medical device companies actively participating in the pilot forgo surveillance, post-approval, and risk-based FDA inspections. Additionally, participants benefit from streamlined submissions for manufacturing change notices, site changes, and PMA notifications.
A primary objective of the Case for Quality pilot is to establish initiatives in collaboration with industry stakeholders instead of simply being known as the enforcer. It is about enabling and encouraging partnerships between FDA and medical device companies.
As most can imagine, this has been met with some degree of reluctance, since historically, FDA and industry professionals have not had this type of mutually serving relationship.
In an effort to drive some of the necessary changes within the regulatory space and to facilitate continuous improvement along with the pilot program, FDA made several adjustments to their engagement activities and submission requirements for participating companies, in addition to the aforementioned benefits.

Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
These changes were intended to reduce the burden and disruption of inspections, while accelerating the review and approval processes, allowing for newly available resources to focus on innovation and improvement.
Value Derived Across Industry Stakeholders
FDA looked at stakeholders across the board to see how the Case for Quality program would deliver the maximum amount of value. For regulatory officials at FDA, 30-day notices for class III PMA products consumed anywhere from 15 to 22 FTEs.
Using this general data, this new program would allow for the agency to significantly increase the number of available resources able to contribute to program improvements. There were also many noteworthy findings in the data gathered among manufacturers.
Under the previous system, product submissions were being limited due to lack of regulatory resources. Some companies were even in the position where they were holding back innovations and ideas because of certain constraints of interactions with FDA.
Comparatively, manufacturers selling into the European Union marketplace were able to move a lot faster through these processes. With public health outcomes in mind, it’s easy to see the importance of lowering those barriers.

Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
The basis for the maturity appraisal model comes down to the practice – how is the work being done? The framework looks beyond just the regulations – actually, it does quite the opposite. The maturity model appraisal embraces how the work is being done by the company; it’s a very interview-driven process.
This model does not hinge on collecting evidence and instead uses a softer approach. It can be understood as a more robust inspection and is designed to drive internal conversations about improvement.
What is Involved in The Voluntary Pilot Program?
We’ve discussed much of what the voluntary program is, so now it’s time to look at some key performance indicators of the program itself. You can check out the application site to find FDA’s criteria for acceptance.
FDA has a primary goal to get the objective metrics possible from this program. There aren’t any metrics in particular that it is seeking, but it is trying to understand the landscape of metrics as it applies to companies.
When an appraisal takes place, it is conducted by a third party. There is some document review – but no document collection. Appraisers work with organizations to understand their objectives so they can determine the best possible avenue to achieve those. In this way, the appraisal becomes a value-add to the organization.
The information collected, how the organization receives from that collected data, and what is reported to FDA is shown here:
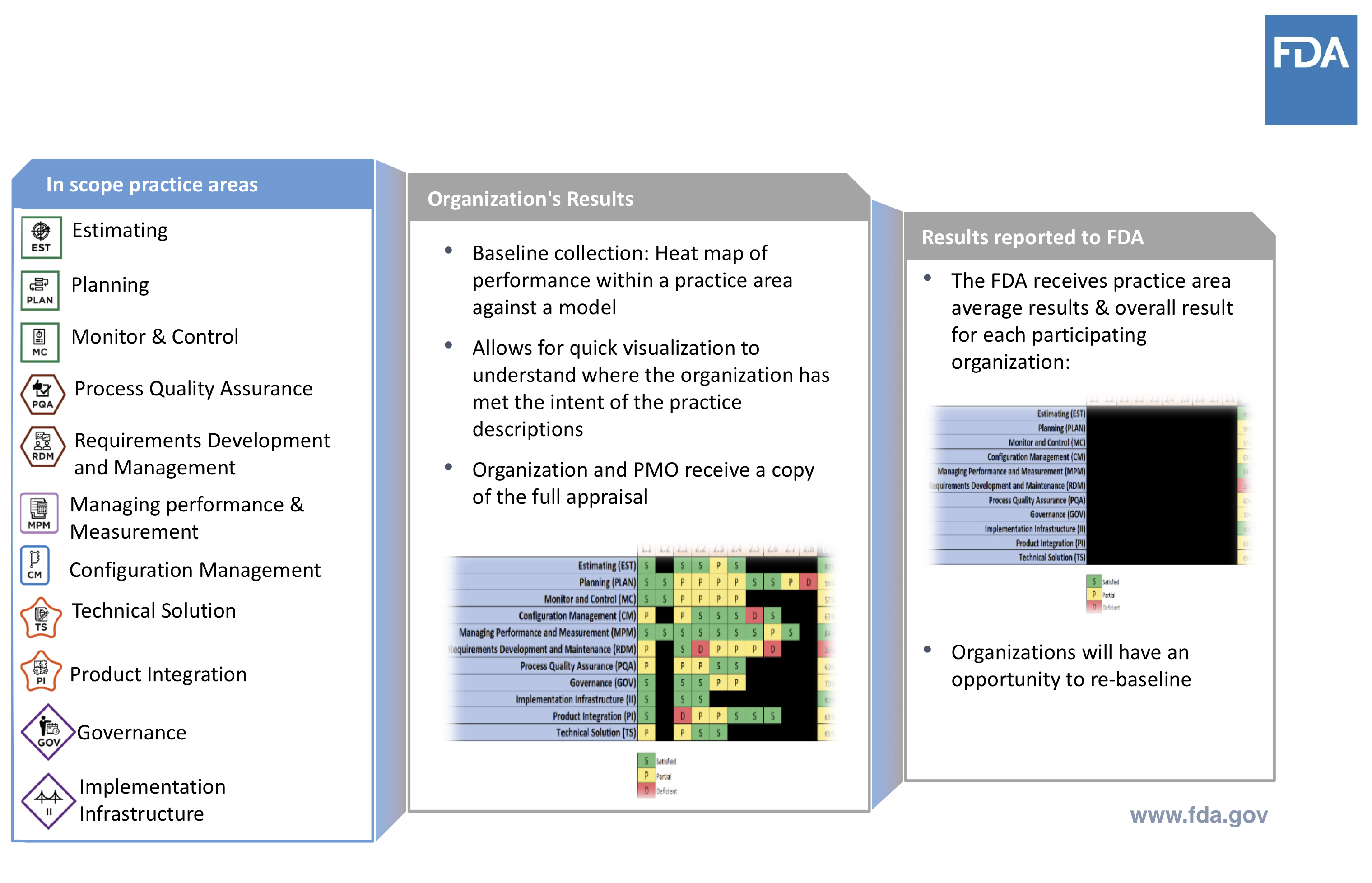
Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
FDA uses this to gather baseline data and make note of any shifts as the company moves through the pilot.
Key Objectives of Case For Quality
FDA wants to have true visibility and insight as to what’s happening within the industry, in any specific product space. This means shifting how it analyzes the information it has collected, with an aim to be more objective.
Determining the least burdensome approach is also high on the agenda. The exchange of information needs to be improved while processes need to be streamlined and error-proof. Underscoring all of this is the desire to accelerate improvement and innovation, leading to better patient outcomes.
While the pilot has focused on manufacturing companies, in the future, FDA would really like to expand to the design sector of the industry. It recognizes that many of the same principles apply in the 510(k) product space, and it needs to understand how to enable that for future Case for Quality initiatives.
A major goal is simplification, which includes aspects of increasing adoption of technologies that facilitate quality, and how to lean out validation efforts.
There’s a current effort on streamlining and clarifying expectations when it comes to non-product computer systems validation. FDA recognizes the need for improved communication of these expectations, after finding a lot of tools had not being adopted due to their perceived regulatory burden.
The use of these systems has the potential to deliver high-value to organizations for improving automation and operational efficiency, directly impacting product and process quality. Identifying what it means to have a quality vs. a compliance mindset is an important part of the equation. This is the focus of the next chapter in our Case for Quality series review.
Part II: Why The Case For Quality Program Matters
With an initial regulatory focus that appeared to be heavily reliant on addressing compliance, many medical device companies have historically viewed FDA as more of a gatekeeper and policing agency, rather than a collaborative partner.
CDRH’s vision is about ensuring patients in U.S. have access to high-quality, safe, effective medical devices. It’s all about the patient and driving best health outcomes possible. With a focus on patient safety and improving quality of lives, the Case for Quality helps shift from being merely compliance-oriented to what truly matters, quality.
By being compliance focused, however, it seems as though what is best for patients can easily get lost.
FDA has been observing several industry behaviors:
- High industry focus on regulatory compliance instead of adopting best quality practices
- Manufacturers not adopting automation and digital technologies
- Little to no competitive market around medical device quality
These observations are what has prompted the need for Case for Quality.
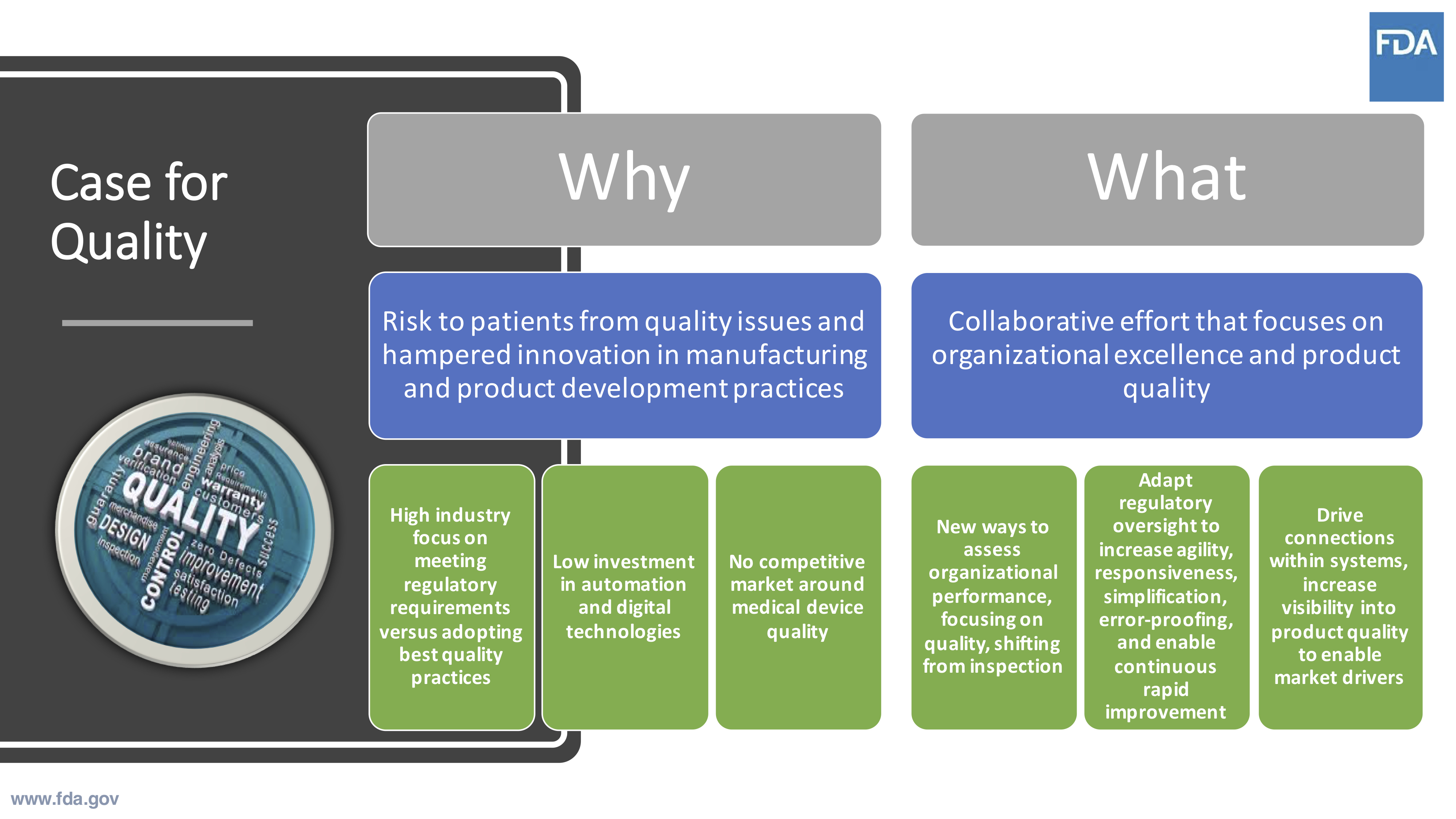
Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
Case for Quality is about driving better integrations within a company’s business systems and processes to understand why the work is being done in order to drive desired outcomes and results.

Shifting From Compliance to Operational Excellence
In addition to improving patient outcomes by focusing on true quality, there are additional benefits for both FDA and industry stakeholders regarding their resource constraints. FDA benefits from reduced load on CDRH resources and FTEs required for inspections and reviews of submissions.
Medical device companies benefit from improving product and process quality. Companies also benefit from reduced internal resource strain currently required to prepare and support submissions and compliance-based inspections.
All of this is for the purpose of getting products to the patients who can benefit sooner. Another side benefit is that reducing time to market for companies will reduce operational expenses and result in revenue realization sooner.
As an example, FDA has seen real benefits with company collaboration already, even in the pilot phase, as it relates to streamlining PMA 30-day notices. As I touched on earlier, there were 30-day notices that might not have been pursued due to resource costs and constraints.
With the adoption of the new model used in this program, these 30-day notices were able to be streamlined, reviewed, and approved within a much shorter timeframe while reducing resource strains for both FDA and industry. As a result, there has been a direct patient benefit.

Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
The current regulatory focus revolves primarily around a company’s quality system. However, running a medical device business successfully requires more than just this. There is a critical need to have a more holistic view of what happens within an organization.
How are tools and resources implemented? What are the company’s values and principles? What are the company’s capabilities and how do these align with their culture, behaviors, and results?

Creating a Virtuous Cycle of Improvement
How is the Case for Quality model truly different from the traditional way FDA assesses medical device companies?
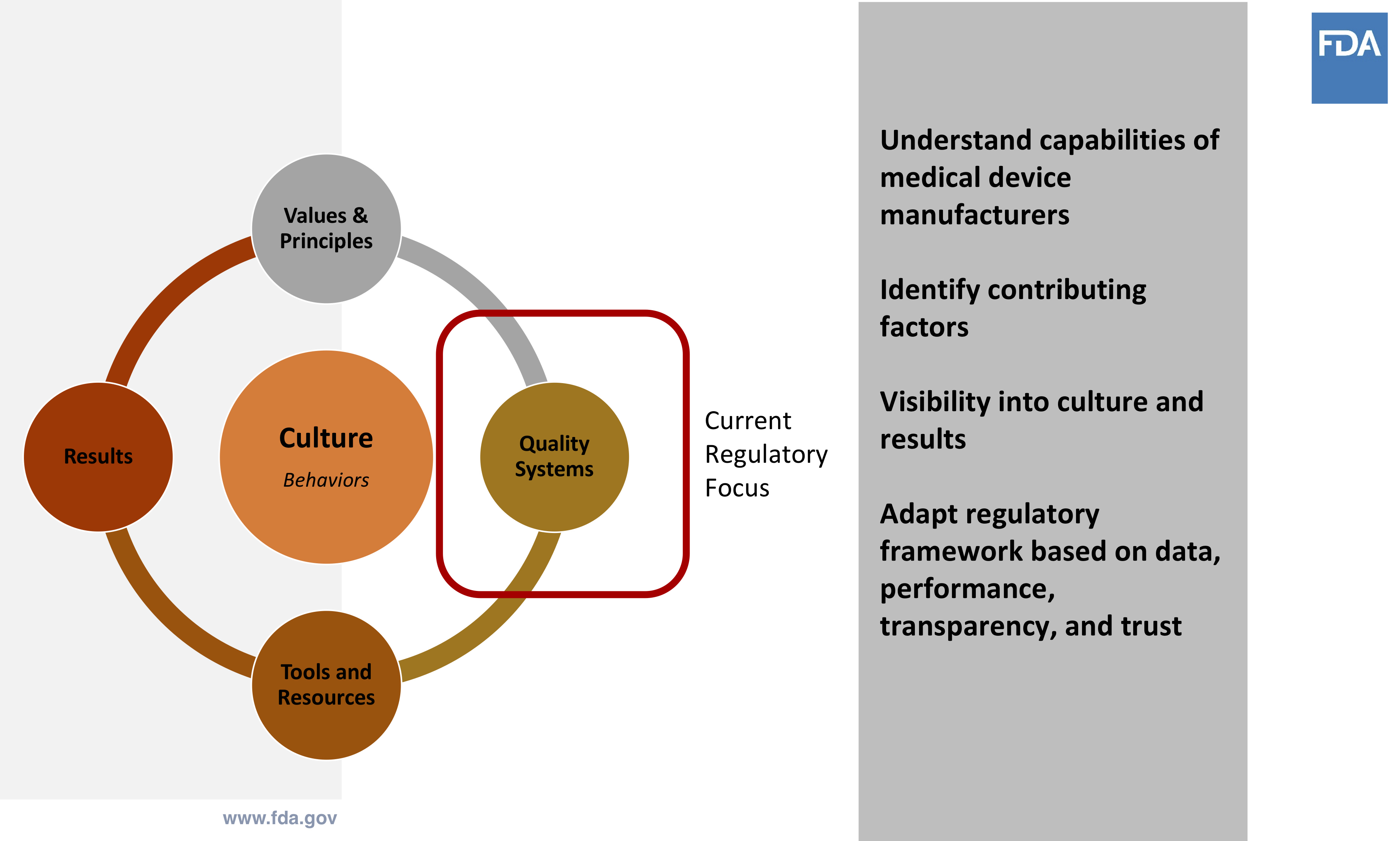
Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
The first step in this cycle centers on understanding the capabilities of the medical device company. This involves a series of interviews to understand the business. This process also involves all relative stakeholders — not just those who handle audits and inspections. The interviews conducted are not an interrogation or typical compliance-based inspection.
The purpose of these interviews is to identify areas for improvement for the company. It’s intended to help enable a company’s ability to be more proactive, rather than just reacting to correct problems.

The Capability Maturity Model Integration
The model this leverages the Capability Maturity Model Integration (CMMI), is not a quality system or a standard. It is a set of best practices that determine a business framework and baseline to assess and evaluate capabilities to more easily identify opportunities for improvement.
CMMI Institute offers a solution that assesses strengths and the framework of the quality management system (QMS) that has been established by the company. Part of the objective here is to assess how well the QMS is performing and whether it is achieving the intended results.
Note: FDA QSR compliance is a prerequisite for inclusion in the pilot. But involvement in the pilot allows a company to forego routine compliance inspections while being part of the Case for Quality initiative.
The Case for Quality initiative has also been driving improvements in FDA processes as well. Some examples include reducing manufacturing change notice reviews from 30 days down to 5 days, manufacturing site change reviews down to a 1-week target, and improvements to PMA processes.
As a result, the device industry is a direct beneficiary. The early results from the program have shown a reduction in FDA review times related to key manufacturing related items, as well as reducing the burden and disruptions of inspections.
FDA envisions future opportunities with Case for Quality, including the ability to help accelerate review and approvals required for product changes, leverage this same methodology for design and development — including the inclusion of 510(k) reviews, and lastly, freeing up resources to focus on continuous process improvement and innovation.
Implementing The Maturity Appraisal Model
How does a medical device company participate with the Case for Quality program? There is an application process that includes CMMI Institute,
a third-party resource to help provide objectivity and to perform the appraisal assessments.
After acceptance, an appraisal team starts working with company. This involves a deep dive into the existing processes at the medical device company to understand current state and desired outcomes. From there, it’s about collaborating and learning to drive quality initiatives.
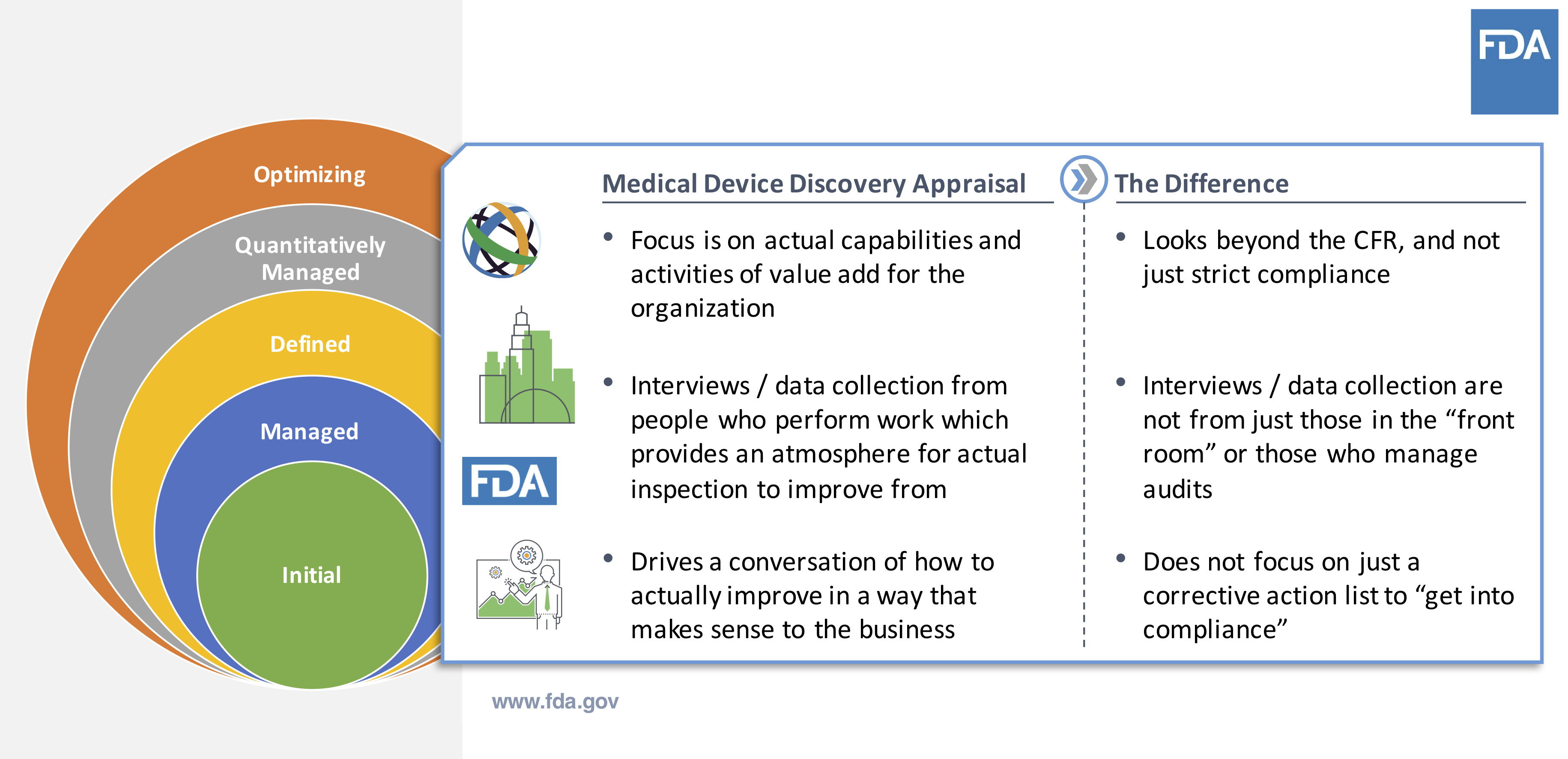
Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
Note that there is no need to do extra preparation to engage in the process. The first steps are about discovery and engaging in conversations to understand current state of the company.
This helps define the existing baseline for the company going through the Case for Quality. To reiterate, this engagement is not an audit, it’s not a FDA inspection.
Case For Quality Program Process
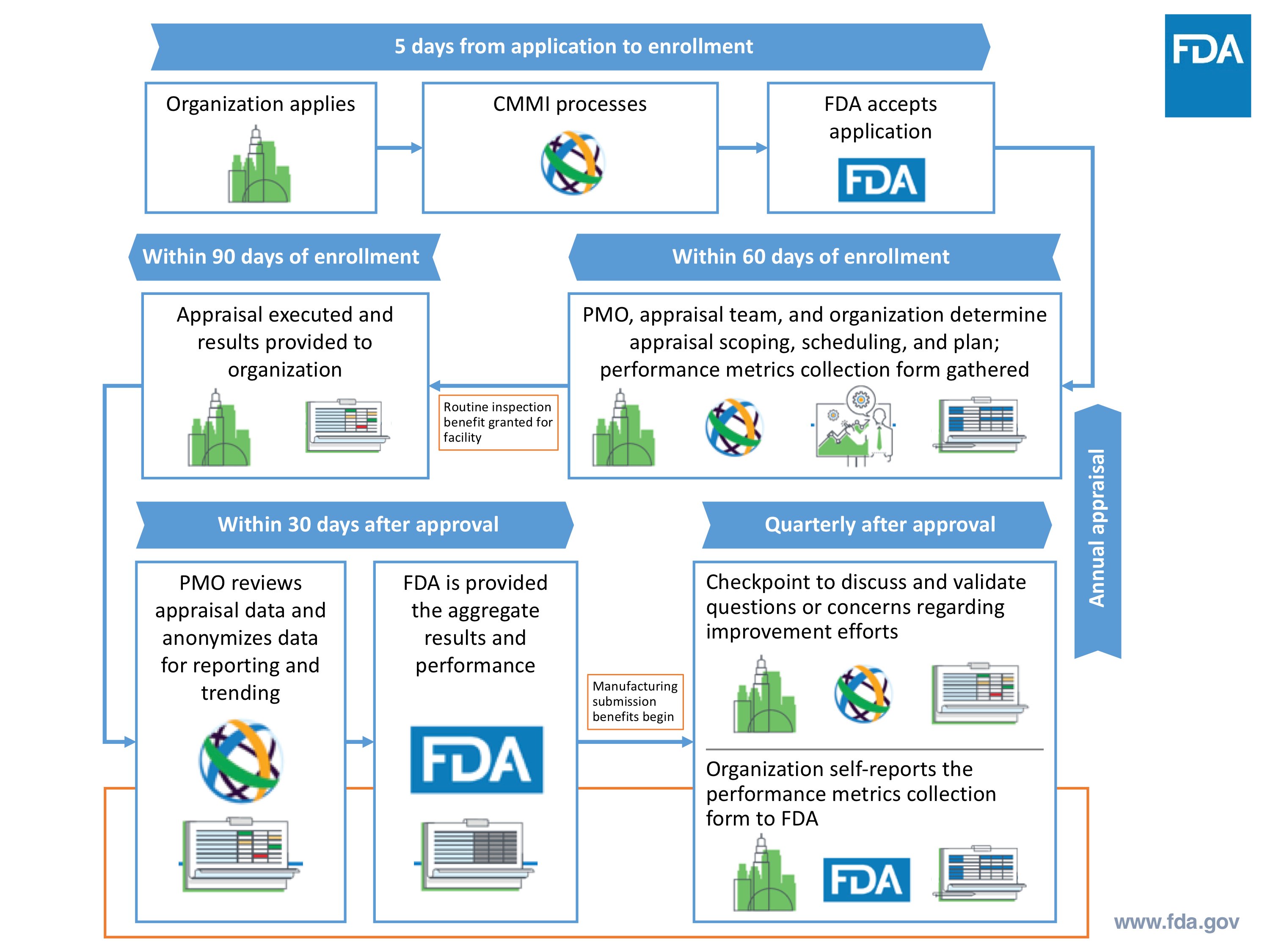
Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
After completing the initial appraisal assessment, the medical device company is provided a detailed process capability report to identify strengths and weaknesses. This is very granular and specific to their business and is prepared by CMMI and visible to the company, but not FDA.
Rather, a high-level overview is provided to FDA in order to establish the baseline of company practices. Recall that FDA already knows that compliance criteria has been addressed because this was part of the inclusion criteria into the pilot program.
Collection of Information
The appraisal assessment is a means to help identify better systems and approaches to identify opportunities for product and process improvements. The methodology is about being proactive versus reactive and to address real problems immediately.
The premise of the entire Case for Quality initiative is to simplifying FDA review processes. It is a means to embrace agency / industry collaboration and provide a format and conduit for meaningful information exchange with the desired purpose for better patient outcomes.
The Case for Quality program aligns with CDRH vision. Frankly, it aligns with the missions of many medical device companies — improving quality of life.
Yes, there is a fear that some companies have raised about this model and approach. Fear that FDA will have access to and use this data and information to identify compliance observations and to provide evidence for downstream inspections.
However, FDA’s overall intent is to eliminate a defensive nature of a traditional compliance-based FDA inspection and improve collaboration to improve patient outcomes.
It has been stated by CDRH, including Dr. Jeff Shuren, there is an interest to remove the term “compliance” from FDA’s lexicon over the next few years. Compliance and enforcement have been synonymous for far too long. Driving focus on quality is about what is best for patient.
Compliance should not be the go-to approach / tool to do this. Those participating in the pilot study to date have indicated that the quality focus is welcomed, and compliance is still being addressed in a healthy manner.

Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
Keep in mind that this Case for Quality approach is new to the medical device industry. And with that, there are some perceptions and assumptions of traditional compliance-based FDA inspection applying to Case for Quality CMMI appraisal. Know that these are different approaches.
With Case for Quality, appraisers have discussions with the actual people who are doing the work within the organization. It’s not about collecting documents and records. It’s about assessing process capabilities. It’s about having real, engaging conversations versus just answering questions.
FDA Inspection vs. CMMI Appraisal
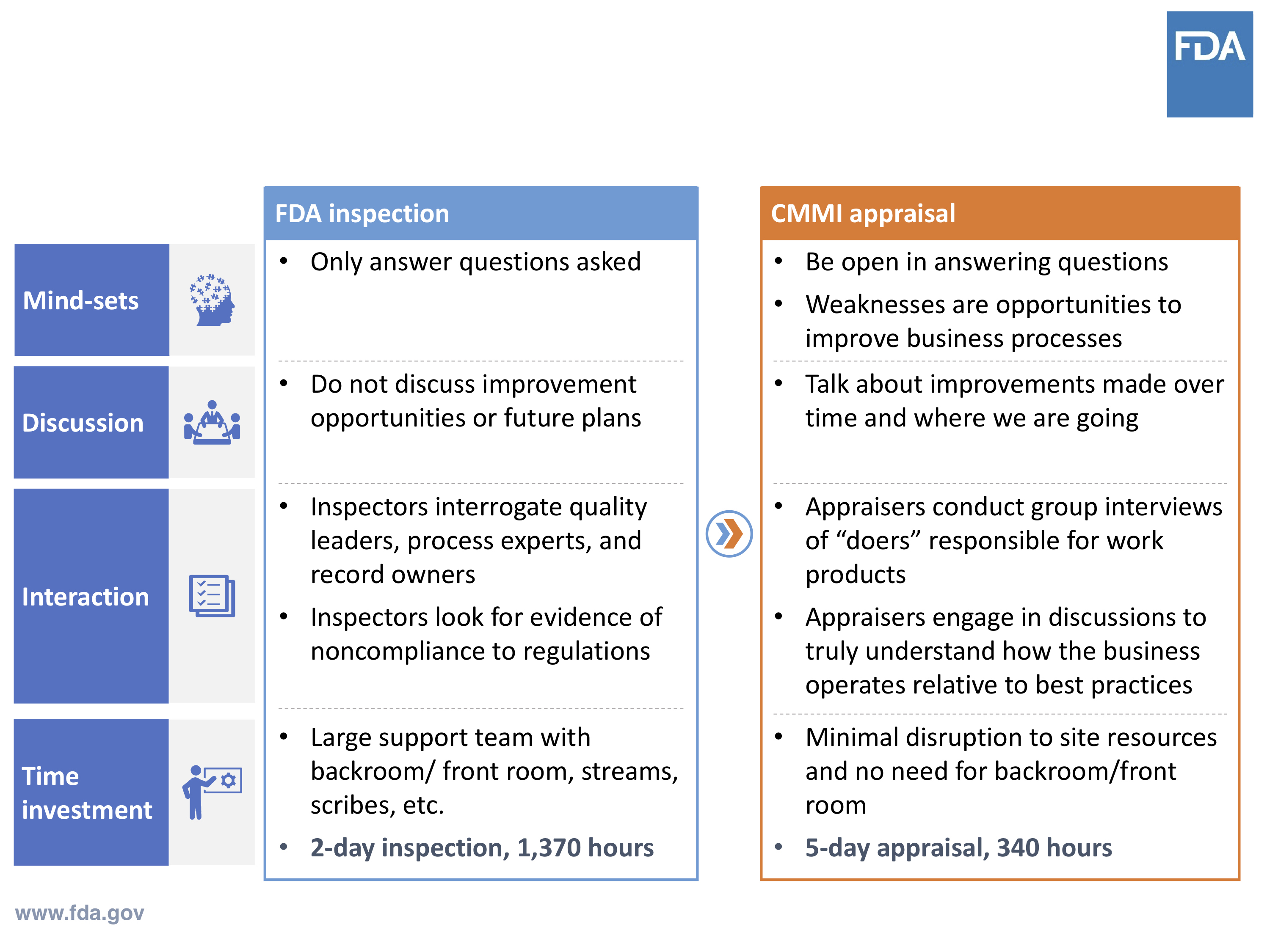
Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
Part of the result of this process is participating companies receive very detailed, granular data and information about their current products and processes. This is key to understanding current state and identifying opportunities for improvement.
FDA receives high-level overviews of each pilot participant, allowing a baseline of quality to be established for the company. This provides the agency more insight into trends and patterns of specific areas relating to quality system effectiveness and current compliance initiatives.
Additionally, this streamlined approach is very beneficial to FDA to better utilize resources and to simplify change notifications and reviews. The Case for Quality provides a framework to better analyze data, drive improved quality metrics, identify areas for more efficient resource allocation, and understand industry needs.
Improved quality metrics are a desired outcome of the pilot program. Historically, metrics used by both medical device companies and FDA are more traditional compliance-based indicators versus quality-based metrics. And this is one area of the Case for Quality that has been more challenging to make the shift from compliance oriented to shifting to true quality.
Compliance Indicators vs. Quality Indicators
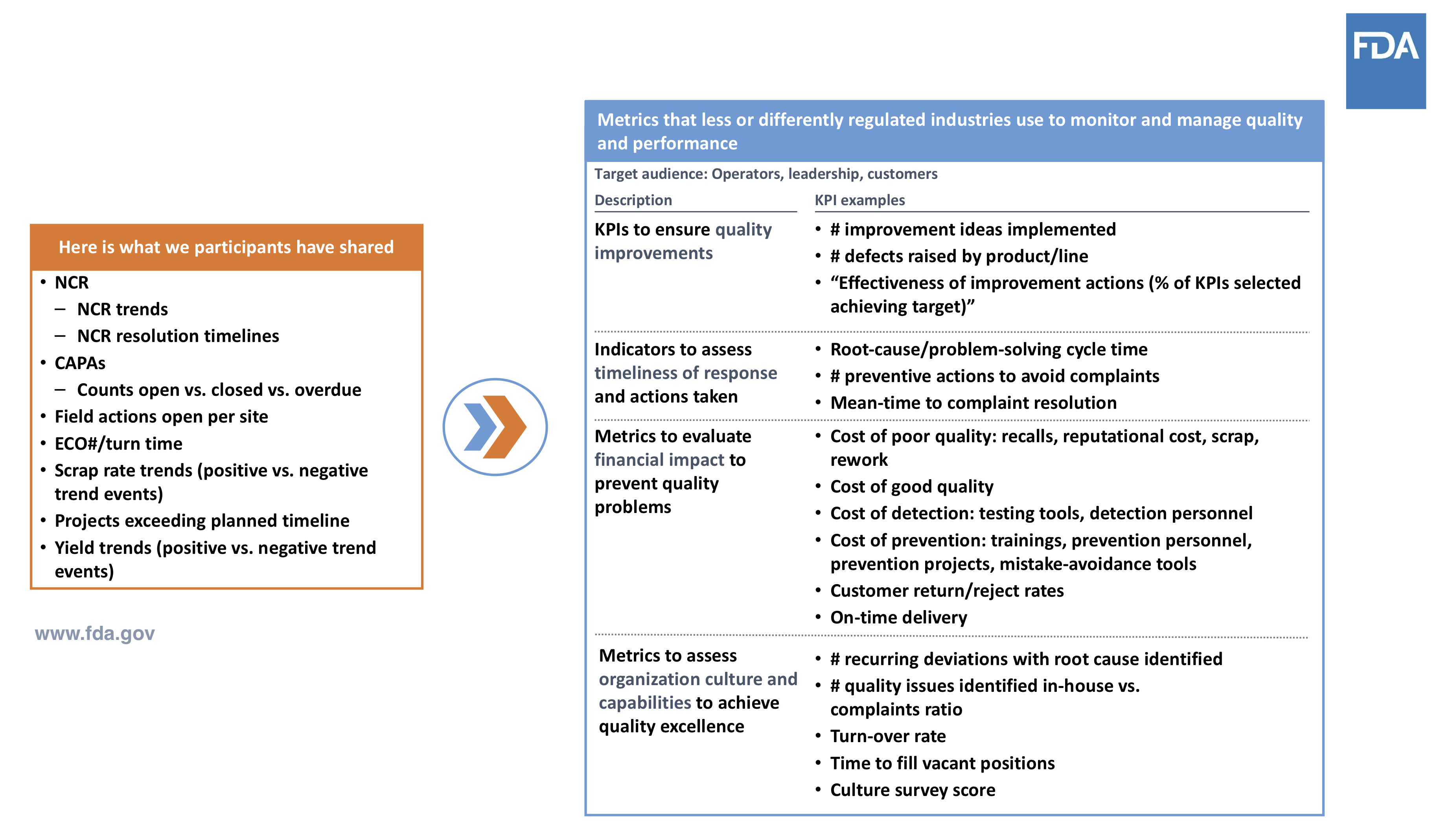
Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
While there are cases for the traditional compliance indicators, shifting the focus to quality-based metrics will have a profound impact on patient outcomes. By having metrics for product safety, effectiveness, reliability, and availability, it will allow for quality KPIs to become front and center.
Ultimately, the goal is to drive alignment between FDA and the company — so that both are speaking the same language with the same overall objective focus on patients. In order for this shift to be possible, it is necessary to have transparency and effective communication between industry and the agency to achieve this alignment.
How do we measure this approach to quality vs. compliance? The metrics necessary to do so are the focus of our next chapter.
Part III: Compliance vs. Quality Metrics
One of the big focuses of the voluntary pilot is the idea of driving improvement across the industry – both for manufacturers and regulations.
According to Cisco, FDA’s role isn’t to hammer on all the areas that aren’t yet green; instead, what they’re interested in seeing is the journey, that there has been consistent improvement over time.
Analyzing Program Metrics
Cisco draws on an example of an assessment result, shared by a company, and how certain processes of theirs have changed over the course of participating in the pilot.
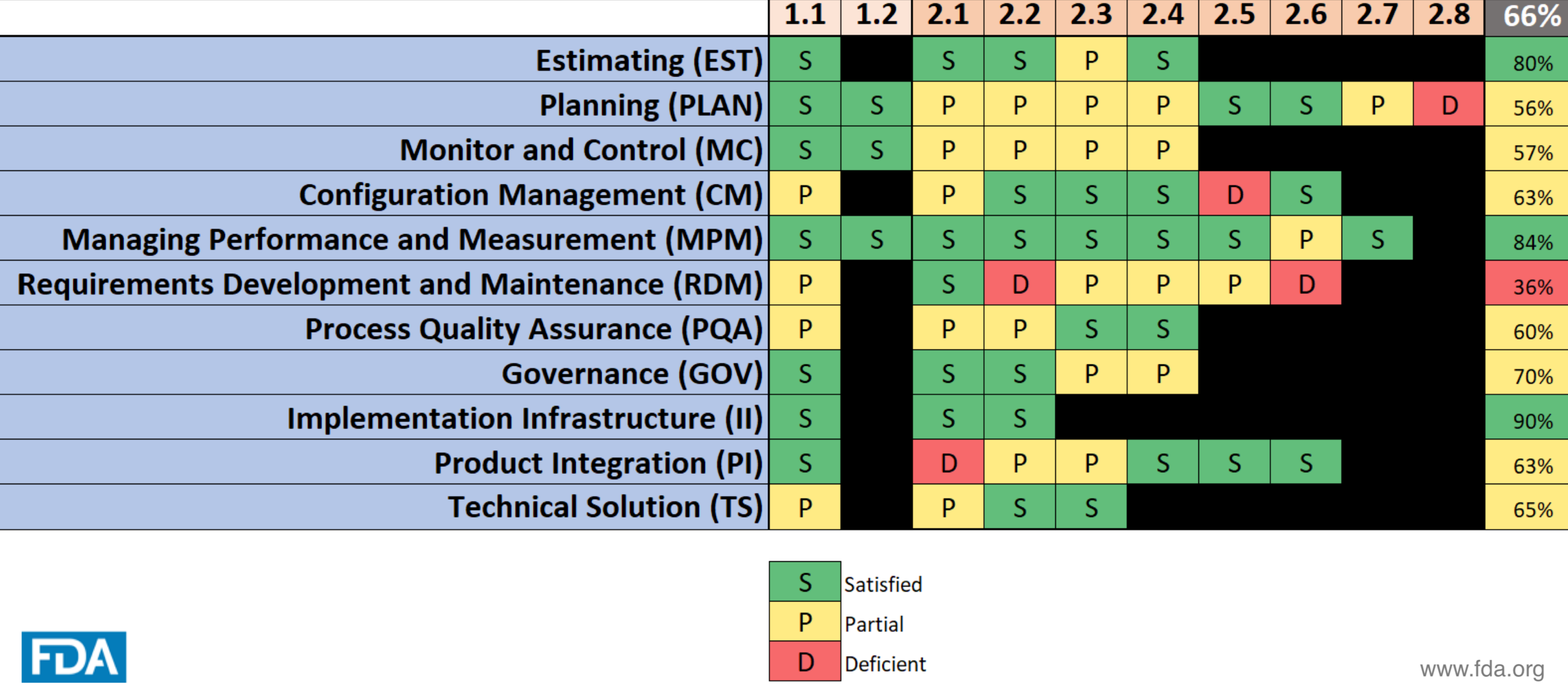
Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
This particular company, whose results are shown above, was able to drive improvement across at least 7 key areas over the course of the pilot period. This idea of driving improvements from a systemic point of view paid off for multiple parts of this company’s system.
FDA is collecting updated data points across all participants in the voluntary pilot, resulting in much better visibility into what’s working and/or not working.
It’s much easier to see whether there’s a recurring problem for a particular manufacturer, as well as problem patterns across multiple manufacturers. This transparency offers a more objective vantage point to extrapolate the data to determine why certain problems exist.
There has been a considerable amount of time spent by FDA delving into its systems and processes to decipher whether there are certain processes not working as well as originally intended. On the other hand, manufacturers are able to get better insights from their end to identify any outliers present that may be negatively impacting operations.
FDA has performed these checks across a broad selection of different manufacturer types and sizes throughout the pilot. The agency plans to carry on with these efforts in order to acquire valuable insight for continuous improvements.
In terms of learning experiences, FDA is seeing that, from a systemic approach, there are things that are not working as well as they would have intended. Some named examples would be CAPA procedures and systems of measurement.
FDA is looking at how and why it gets the information it does. Is there a better way or a “least burdensome” approach? More importantly, can it achieve the outcome it is looking for (safety, effectiveness) in a faster, more reliable fashion?
Overall it is finding that it can implement more changes in a faster time frame than what it could on an individual review basis. It is also seeing a key mindset change among participants – from a compliance focus to problem-solving and from hesitancy to engagement.
Collaboration and learning have increased, with ideas being pitched from manufacturers and FDA. There has been an evident shift from “assumptions and reacting” to “understanding and insight.”
Compliance vs. Quality Indicators
A key goal of the pilot is to get to the point of measuring and monitoring what really matters. One thing that FDA did with participants from early on is ask them what they thought were important measurements to use.
As Cisco recounts, this process took some trust-building. People wanted to know what they were going to do with that information and what it meant. In the beginning, they were given a lot of “here’s what you want to hear” compliance indicators.
In more recent times, the indicators given by the companies have shifted. For example, more are focusing on employee health and safety as a big deal, which leads to other indicators being looked at within the organization.
In the chart below, there’s a clear comparison of early participant versus more recent performance indicators. Note how the more recent measures given tend to provide a more robust picture of quality than those earlier suggestions.
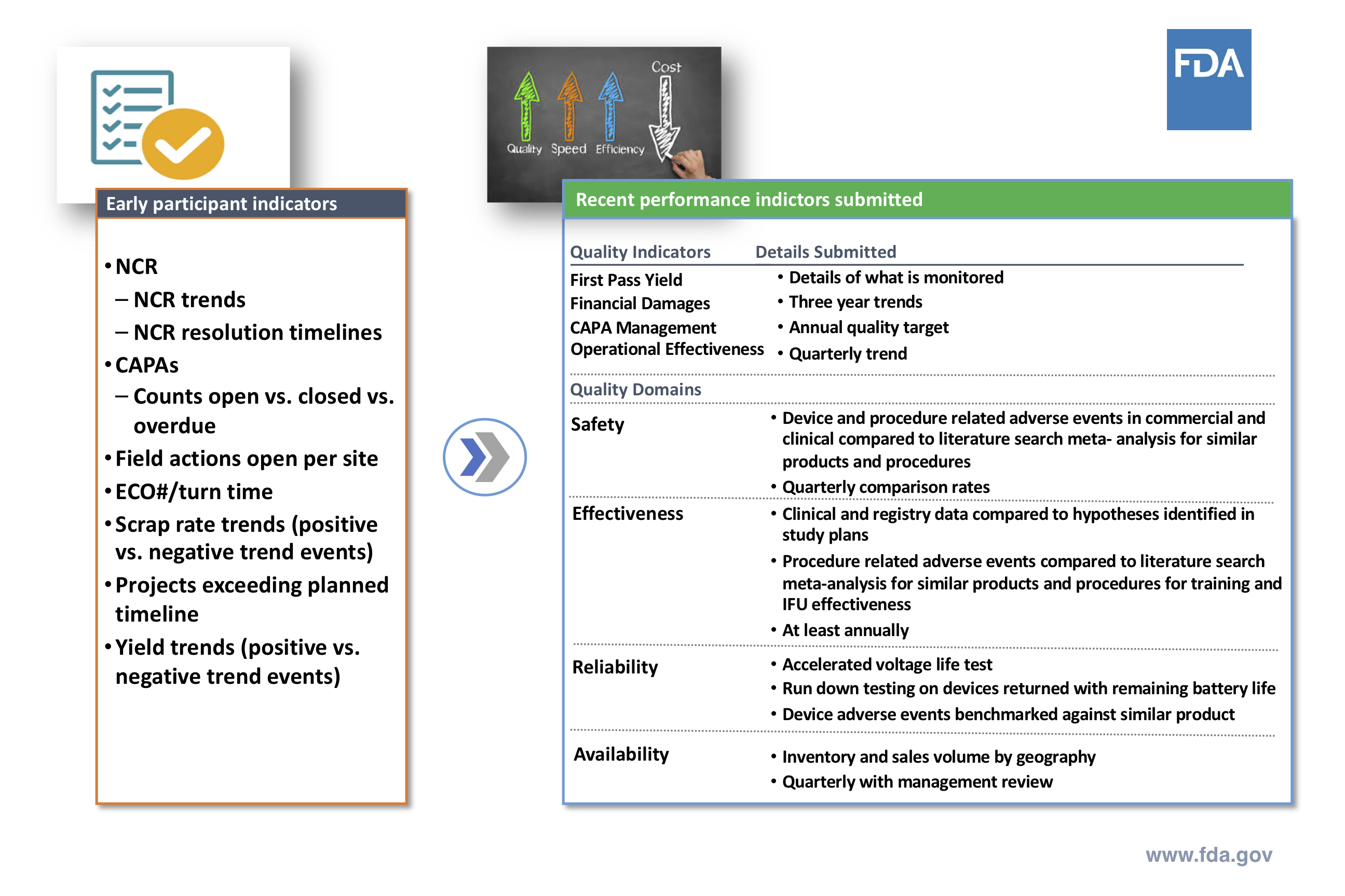
Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
An important metric for FDA is results gained, rather than a company focus on compliance. Areas, such as CAPA and Operational Effectiveness, have shown promising improvements among participants, along with several other areas.
The underlying point is that quality is an overall focus, rather than simply being compliant with an item on a checklist.
Of further note is that they’ve received some great suggestions in terms of quality domain metrics from participants. Metrics covering safety, effectiveness, reliability and availability have been extrapolated on, in order to provide a framework that any medical device company can follow.
Why is there a focus on quality indicators, KPIs and metrics? One of the things FDA is looking to do with companies is being more objective. It wants to see the result of the process so that the focus isn’t on control and fixation of the process.
Enhancing Visibility and Innovation
The goal is that once a process has been established and approved for the organization, they are then able to truly innovate around the process and put more focus on the results that it generates.
This gives FDA a lot more visibility over what is happening within the organization and gives the organization the ability to align the process with their business. The intended shift here is moving from a compliance mindset to a quality improvement mindset.
By adopting this new mindset, companies can address any issues as quickly and effectively as possible. There will be issues that still come up, but a quality system is developed in this process with the intention of being able to account for those potential issues.
FDA would like the aforementioned quality and safety outcomes, but it also understands that a medical device company is a business. How can it work with business and quality needs to deliver better outcomes for both?
The slide below from Vicenty highlights key ideas for why this program is important:

Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
Applying Proven Methods in Future Programs
This current pilot has focused on the manufacturing side, but in the future, FDA would like to expand to design and supplier management. With the proper processes in place, FDA can efficiently replicate the same methods to multiple sectors in the industry to enable speed, responsiveness and overall improvement to their 510(k) process.
In terms of systems, FDA is keen on improving future and existing regulatory system elements. It would like to increase industry adoption of tools, practices and performance measurements, and establish a fully accredited FDA program by 2019.

Breaking down barriers has been an important part of Case for Quality, and one of those identified has been non-product computer system validation. This is the focus of our final chapter.
Part IV: How The Pilot Addresses Non-product Computer System Validation
Cisco Vicenty of the Office of Compliance presented the final webinar in our Case for Quality series where he highlighted all of the work the agency has been doing on Non-Product Computer System Validation.
This is an area FDA identified as presenting challenges to many companies, and it hopes that medical device companies will be able to better leverage computer-based systems in the future.
Breaking Down Industry Barriers
As part of its Case for Quality initiative, FDA has worked closely with medical device companies to understand the fundamental barriers that need to be addressed. Cisco recounted many conversations with companies that confessed that the investment in software and technology hadn’t been a priority for them.
Given that software has improved in leaps and bounds in recent times, FDA took a large interest in understanding why companies would hesitate to invest in these tools. It was discovered that computer software validation (CSV) was seen as a huge hurdle for device manufacturers.
Companies were concerned about the expense involved with validating their systems, as well as the associated regulatory compliance risk. The cost of system validation was in many cases up to two times the amount of the base system cost.
“This was an eye-opener for us at the FDA,” Cisco says. The agency had been encouraging this adoption of new software for quality systems, but had not realized the extent of the financial barriers that existed for manufacturers.
FDA’s Position On Automation Tools
As confirmed by Cisco, FDA supports and encourages the use of automation and that it has the potential to help with better product knowledge, tracking and trending, plus a host of other applications.

Manufacturers can gain advantages from automation throughout the entire product lifecycle. They can reduce or eliminate errors, optimize resources and reduce patient risk.
FDA’s position is that using these sorts of software products can be an excellent way to enhance product quality and safety, which in the end, is the overarching goal.

Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
Cisco describes this opportunity to improve as “low-hanging fruit” for both the companies and FDA. A wise investment in automation tools can also help to boost the overall value of a business. It’s very much up to companies to make the decision out of the many choices available.
Streamlining Validation Processes
Part of the problem with CSV that was found by FDA had to do with a lack of understanding of the process and the exclusive focus on compliance from manufacturers. Companies were so worried about just being compliant that they didn’t want to add another layer of risk to the computer system.
Overall, it was concluded that the medical device industry is significantly lagging with regards to the implementation of automated systems and new technology. Perceived regulatory burden and outdated compliance practices have reduced abilities to learn, react to issues and improve product quality.
Given the current state of the process for software validation, FDA recognizes the need for a paradigm shift to more value-driven and patient-focused approaches. The regulatory agency advocates for critical thinking and risk- based, agile approaches to streamline assurance activity and evidence capture.
What has been happening is that the vast majority of companies’ focus is dedicated to the documentation or regulatory compliance, while critical thinking only comprises a small part of the process. FDA would like to see this flipped on its head.
Cisco revealed that a review of FDA’s software guidelines is on the books for 2019 in the form of a specific guidance document for device manufacturers. As he also pointed out, FDA opted for a guidance rather than a review of the regulatory language because there is nothing there right now stopping companies from taking this approach.
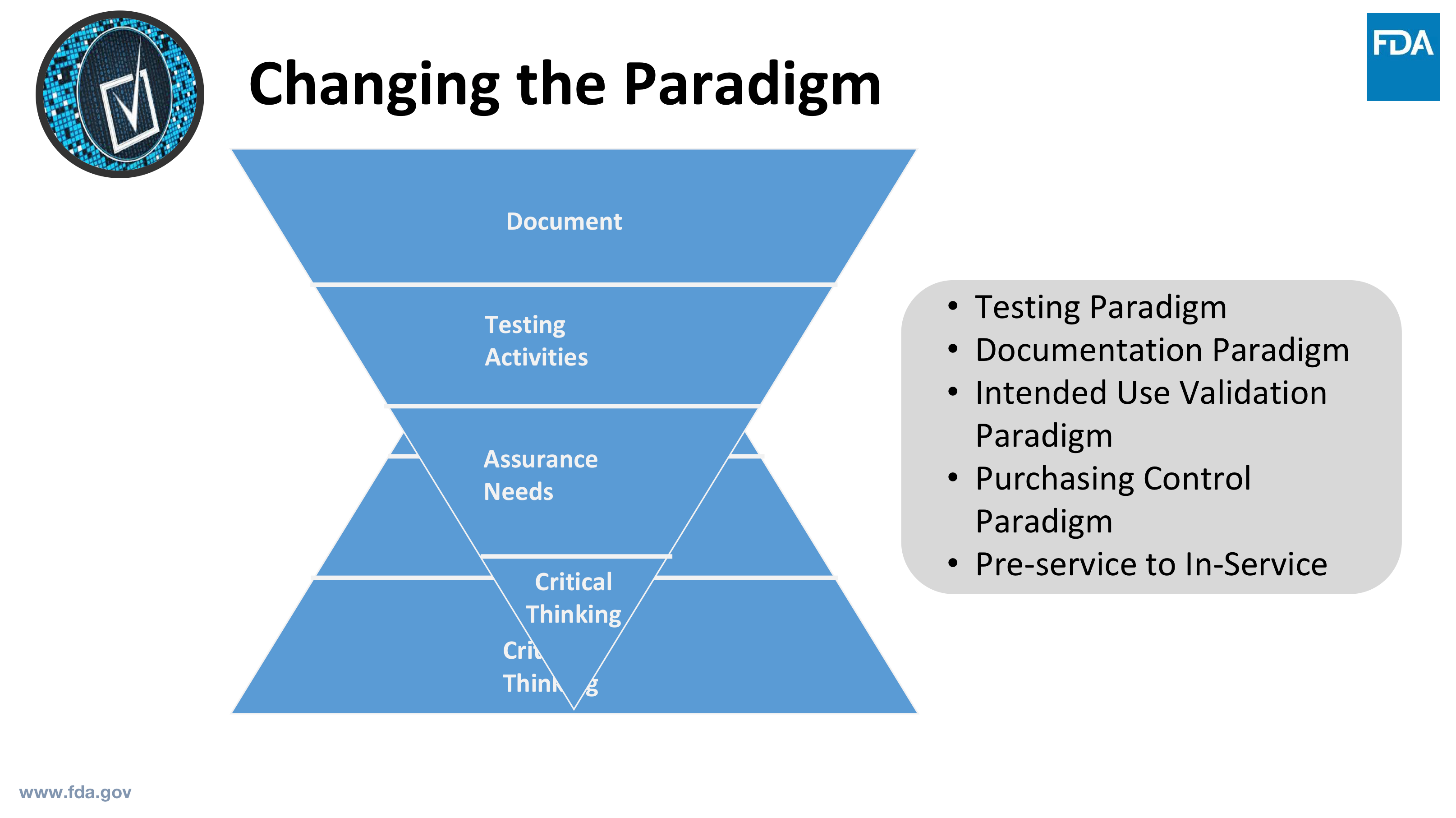
Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
Changing The Paradigm
One of the first things Cisco believes can be a catalyst for changing the paradigm is shifting the discussion. Instead of a “did you validate?” approach, we need to start from an assurance perspective first.
What this means is that instead of the conversation always revolving around system verification and validation, the conversation would sound more like, “how are you sure that this meets your needs?”
What does computer software assurance look like to FDA? Cisco breaks it down into a process that begins with identifying intended use. Does the feature, operation or function directly impact device safety, quality, or your quality system integrity?

You can then determine a risk-based approach, and as Cisco points out, “there is nothing in the regulations preventing you from doing this. Will this directly impact device safety? If so, take this a step further: will this cause harm to a patient?”
From there, Cisco talks about the methods and activities for assurance (shown in the slide below) and the appropriate record of these activities. He points out that it is up to the individual company to determine what is least burdensome.

Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
The documentation made throughout this process needs to be of value to you as an organization, not whoever is coming in to audit. You do not need to have hundreds of pages for it to be valuable.
Impact on Quality Assurance Landscape
FDA does not intend to focus regulatory resources on inspection of quality assurance activities, as they could be better focused elsewhere.
Wherever there is a direct impact to the device quality or safety, the manufacturer is responsible for identifying the features or functions causing the impact. Doing so will enable the manufacturer to capture the associated risk and execute appropriate assurance activities to prove risk was evaluated and the system performed as intended.
FDA will however focus its regulatory activity on inspection and review of those systems which do impact quality or safety. What FDA really cares about is the risk to quality or safety, and how the software has a direct impact on these things. You can see the areas they look at highlighted here:

Source: Cisco Vicenty, CDRH FDA Case for Quality Program Manager
Cisco outlines some acceptable testing methods for software assurance, along with how to record results, which we share in a downloadable checklist.
He points out that currently, companies are often focusing on the most robust testing, adding to the “burdensome” impression of software assurance. It is not always necessary to take this path.
“Always consider the value of the record you are intending to keep, and keep in mind that “least burdensome” principle,” Cisco says.
Automated Computer System Validation Tools
The question of using automated computer system validation tools always goes back to intended use. Cisco says the agency aims to provide some clarity around these in the upcoming guidance.
There are a number of off-the-shelf tools available, capable of delivering great results with full traceability. FDA wants to enable greater use of tools such as these. There are also a number of tools available to manufacturers for testing these systems.
Cisco says FDA does not want to spend a lot of resources and time on reviewing those ancillary tools. He highlights that they’re always going to go back to intended use.

The manufacturer is using these tools to automate and supplement the tracking and assurance testing for their non-product systems. The intended uses of these tools do not have a direct impact on device quality and device safety.
The Future of Case For Quality
With the success of the pilot in 2018, the natural question is, what’s next? CDRH Center Director Jeff Shuren announced at MDIC’s Annual Public Forum in September CDRH’s intention to move the pilot to a full program in 2019.
CDRH will continue to partner with MDIC, CMMI Institute, and other industry partners to expand the pilot. It’s important that the program continues to meet FDA and industry needs to deliver safe and effective products for patients.
MDIC will work with CDRH on program oversight, building a program that continues to encourage mature quality practices. The pilot was intentionally designed with the flexibility to expand regulatory incentives and add additional practice areas.
As MDIC continues to work to advance the Case for Quality, new initiatives are underway to redesign the Corrective and Preventive Action (CAPA) process as a continuous
Additional work streams have been established to encourage the development of university curriculums on quality and develop tools for engaging senior management teams to drive the quality discussion across their organizations.
Collectively, these programs and initiatives will help support the vision of the Case for Quality -- to move the medical device ecosystem from a mindset of compliance to a broad culture of quality.

Final Thoughts
This concludes our series on FDA’s Case for Quality Pilot Program. It has been a truly collaborative effort that would not have been possible had it not been for the contributions and support from our esteemed partners, FDA and MDIC.
We’d also like to express our gratitude to Cisco Vicenty at FDA for his participation as the keynote Presenter for Greenlight Guru’s webinar series. We look forward to seeing further shifts in the approach to quality.
If you have any further questions, feel free to contact us directly and we’ll be happy to answer whatever we can. The full 4-part webinar series is available free and on-demand, with exclusive Q&A sessions from Cisco and audience attendees about Case for Quality and why it matters.
The Case for Quality program has proven to be a catalyst for a systemic shift in how manufacturers and FDA interact with one another. A new collaborative environment was born where quality was a primary emphasis over mere compliance, which has laid the foundation for a landscape of industry improvements and success.
MDIC will continue to work closely with the medical device industry and CDRH to advance the Case for Quality initiatives. Its main objective is to encourage an industry ecosystem that moves beyond compliance to high-quality manufacturing and products. You can also find additional information about the Case for Quality and information on how your company can get involved.
We’re looking forward to seeing more from this program, and the continued initiatives outlined for 2019. At Greenlight Guru, helping medical device companies focus on quality is our goal, so the FDA Case for Quality program aligns very closely to our company’s foundational core values. We look forward to serving as their Quality Management System affiliate and collaborating on planned future initiatives.
About the Authors
Greenlight Guru
Greenlight Guru is the leading Quality Management Software Platform designed specifically to serve the medical device industry. Our cloud-based solution is being used by device makers in over 600 cities and 50 countries to bring safer devices to the markets they serve, faster while reducing risk.
With FDA/ISO guidelines built into workflows within the software, companies can achieve end-to-end traceability, automate and track activity, and improve the quality of any submission. Our industry-specific solution allows teams of all sizes to maximize productivity and streamline processes throughout the entire design and development lifecycle.
In addition to our software, Greenlight Guru also offers in-house QA/RA Services. This white glove service is led by our team of Gurus, who have 10+ years of industry experience and are committed to working with our customers to implement and advance quality culture.
Jon Speer is the founder and VP of QA/RA at Greenlight Guru and the main contributing author of this publication. Jon is a medical device industry veteran with over 20 years experience having helped dozens of devices get to market over his career in a variety of roles including product development, project management, quality and regulatory. He is a thought leader, speaker and regular contributor at numerous leading industry publications. He is also the host of the #1 most downloaded podcast in the industry, The Global Medical Device Podcast.
Medical Device Innovation Consortium (MDIC)
The Medical Device Innovation Consortium (MDIC) is a public-private partnership collaborating on regulatory, scientific, and health economic challenges within the medical device and diagnostic industry.
Through its partnership with industry stakeholders, MDIC coordinates the development of methods, tools, and resources used in managing the total product life cycle of a medical device.
Offering guidance and leadership, MDIC members shape the future of healthcare by providing subject matter expertise to working groups aimed at advancing approaches that promote patient access to safer and more innovative medical technologies.
Stephanie Christopher, MA, has a background in health communication, public health project management and communication education and training. Stephanie joined MDIC in 2013 and manages MDIC’s patient centered benefit- risk assessment, patient engagement and quality initiatives.
Prior to joining MDIC, Stephanie worked with an academic public health team working on interventions to improve the quality of communication between physicians and parents of newborns with abnormal newborn screening results.
In 2012-13, Stephanie went on leave from her academic position to do a special assignment for the Food and Drug Administration Center for Devices and Radiological Health (CDRH), updating and training staff on a new risk communication process. Stephanie has also served as an adjunct instructor at Marquette University, teaching introductory communication courses.
Stephanie earned her Bachelor of Arts in Communication-Print Journalism from Pacific Lutheran University in Tacoma, Wash. and Master of Arts in Science, Health, and Environmental Communication from Marquette University in Milwaukee, Wis.
Stephanie is also a Certified Clinical Research Coordinator (CCRC) through the Association of Clinical Research Professionals and in 2018 was named a Fellow of the Association of Clinical Research Professionals (FACRP).
Leah McConnell is an award-winning communications and marketing director with an extensive background in the biomedical and health sciences field. Leah joined MDIC in 2018 to bring the organization’s work to life through dynamic and compelling stories that engage the med tech industry and the wider community more deeply with MDIC’s mission.
As MDIC’s first Director of Marketing and Communications, Leah provides vision, leadership, and implementation of strategic communications, sustainability branding, and media relations across multi-channel platforms. She is also responsible for developing relationships with key external partners and manages the organizations membership of diverse industry stakeholders.
Prior to joining MDIC, Leah has supported a portfolio of companies and organizations to include projects with U.S. Air Force, Independence Blue Cross, FDA, Naval Medical Research Center, and more.
Leah earned her Bachelor of Science in Communications Media from Indiana University of Pennsylvania and her Master of Science in Public Relations and Marketing from University of Denver. She is a recipient of two Gold AVA Digital Awards for her work with content design and development as well as building successful social campaigns. She is also a recipient of The Power 30 Under 30TM Awards for professional and community excellence.



