
NOTE: The FDA draft guidance on Computer Software Assurance for Production and Quality System Software is now available. For more information, check out our articles on computer software assurance and how the guidance recommends you carry it out.
What you’ve known and believed about computer system validation, software validation, and 21 CFR Part 11 compliance is about to change in a huge way.
And the change will be positive, simplifying this topic of confusion and actually streamlining your life as a medical device professional who would like to embrace automation and technologies within your business.
This past year, Greenlight Guru partnered with the FDA on their impactful Case for Quality program through a special 4-part webinar series, which is now available for download. In the final segment, Cisco Vicenty, CDRH Program Manager at FDA, offered his unique insight on the topic,
The FDA supports and encourages the use of automation, information technology, and data solutions throughout the product lifecycle in the design, manufacturing, service, and support of medical devices.
Automated systems provide manufacturers advantages for reducing or eliminating errors, increasing business value, optimizing resources, and reducing patient risk. These capabilities provide significant benefits in enhancing product quality and safety.
In 2019, FDA will be releasing a new, draft guidance “Computer Software Assurance for Manufacturing, Operations, and Quality System Software” that embraces modern thinking on the topic.
I am looking forward to this guidance because, in my experiences, I’ve made and observed decisions regarding automation and technology that were often hampered by fear and pain of validation.
Let me provide a little background and more context on the topic.
The current FDA regulations pertaining to computer systems is defined in 21 CFR Part 11, and these regulations were defined back in 1997 and unchanged since. The FDA did release its current guidance on “General Principles of Software Validation” back in 2002 and guidance on Part 11 in 2003. And unless you’ve been off the grid for the last 15 years, you know that software development and testing methodologies have evolved significantly since then (for example, consider cloud computing and mobile apps).
Yes, FDA had previously attempted to revise interpretation and guidance on Part 11 and software validation a time or two since then, yet with little avail. The regulations and guidances regarding what is expected regarding software validation has not evolved at anywhere near the same rate.
Why is this a big deal?
Regulatory expectations have been highly influential in how and what medical device companies choose to do with respect to computer systems and software validation. The prevailing conventional wisdom on Part 11 and software validation is that regulations are onerous and overly-burdensome.
So much so, that the majority of medical device companies still today rely heavily on paper and general purpose tools for their documentation and quality management systems, rather than implement software solutions and automation to assist in these areas.
In my honest opinion and based on my decades of experience, Part 11, computer system validation, and software validation have been extremely confusing by all in the industry--medical device companies and regulators alike.
FDA used the term “enforcement discretion” several times in the 2003 Part 11 guidance. What does enforcement discretion mean? And how do you address Part 11 criteria when the guidances are somewhat vague and interpretations are highly varied?
There is a silver lining in the near future regarding this whole topic. FDA has announced that a new, modern draft guidance regarding computer system software, “Computer Software Assurance for Manufacturing, Operations, and Quality System Software”, is on the docket for 2019.
And what I’ve learned from Cisco Vicenty, the Program Manager for FDA CDRH Case for Quality, is that FDA has a keen interest in simplifying this for several reasons, not the least of which are improved product quality and patient safety.
CSV Silver Lining
As part of Case for Quality initiative, FDA is focused on helping medical device companies shift from a compliance mindset to a true quality, metrics-based focused. What FDA learned as part of the initiative is that medical device companies are generally doing a poor job of leveraging automation and technologies that can impact internal systems.
What is it that prevents use of technology?
FDA found that computer system validation (CSV) efforts have been too extensive and too expensive for companies to embrace. The current industry practices and lack of adoption of automation and technology have resulted in numerous unintended consequences.
FDA found that some companies were spending up to two times the cost of software on validation exercises. Yet, these actions have been deemed required due to the focus on compliance. Interestingly, a compliance approach actually has the potential to increase risk to patients. FDA observed that compliance-centric approach has resulted in quality issues and has hampered innovation in manufacturing and product development practices.
This was a wake-up call for FDA.
So much so, that FDA’s current view seems to have shifted (some might say quite dramatically) in favor of encouraging use of automation and technology.
Leveraging automation and technology provides medical device companies with better information, product performance knowledge, easier methods to track and trend, better equipped for more timely responsiveness, process optimization, improved patient experiences, reduction in product risks, reduces operational expenses, and increases business value.
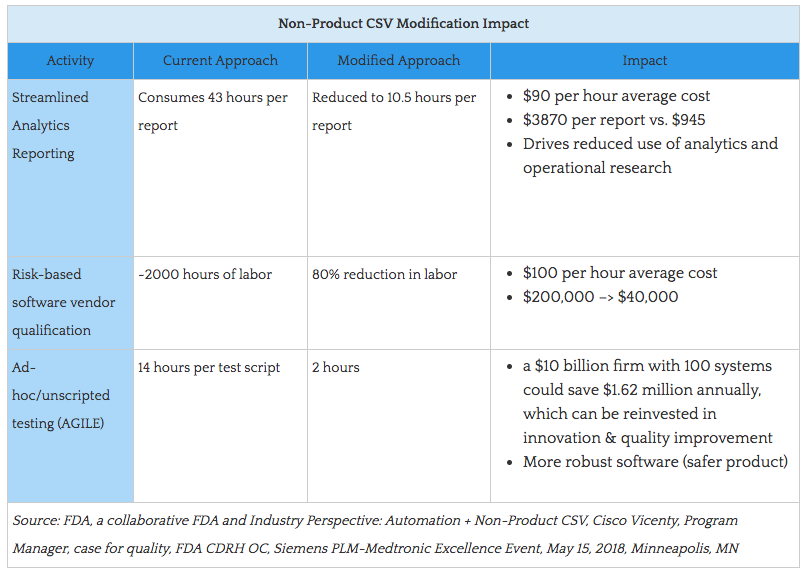
And this shift should be applicable to all types of automation and technologies with the objective to drive best possible outcomes and performance. FDA wants the medical device industry to hear loud and clear:
Do not fear investing in the automation and technology to help you run your business. In fact, embrace it.
Yes, I know. Confusing. Confusing, in part, due to lack of clarity from FDA. Let’s be real. For decades, the industry has been more compliance-focused than true quality focused.
Shift in Validation Paradigm
With the shift in FDA position on automation and technology, a shift in the validation paradigm is also essential.

Source: Cisco Vicenty, FDA Case for Quality Program Manager
It is expected that the new, draft guidance to be released in early 2019 will elaborate and expand on this new approach with CSV. Despite this, realize that this approach is completely supported and doable right now within scope of regulations.
Both the 2002 Software Validation and 2003 Part 11 guidances embrace the concepts of least burdensome and risk-based approaches. However, this is not the approach that most of us have been accustomed to regarding CSV.

Source: Cisco Vicenty, FDA Case for Quality Program Manager
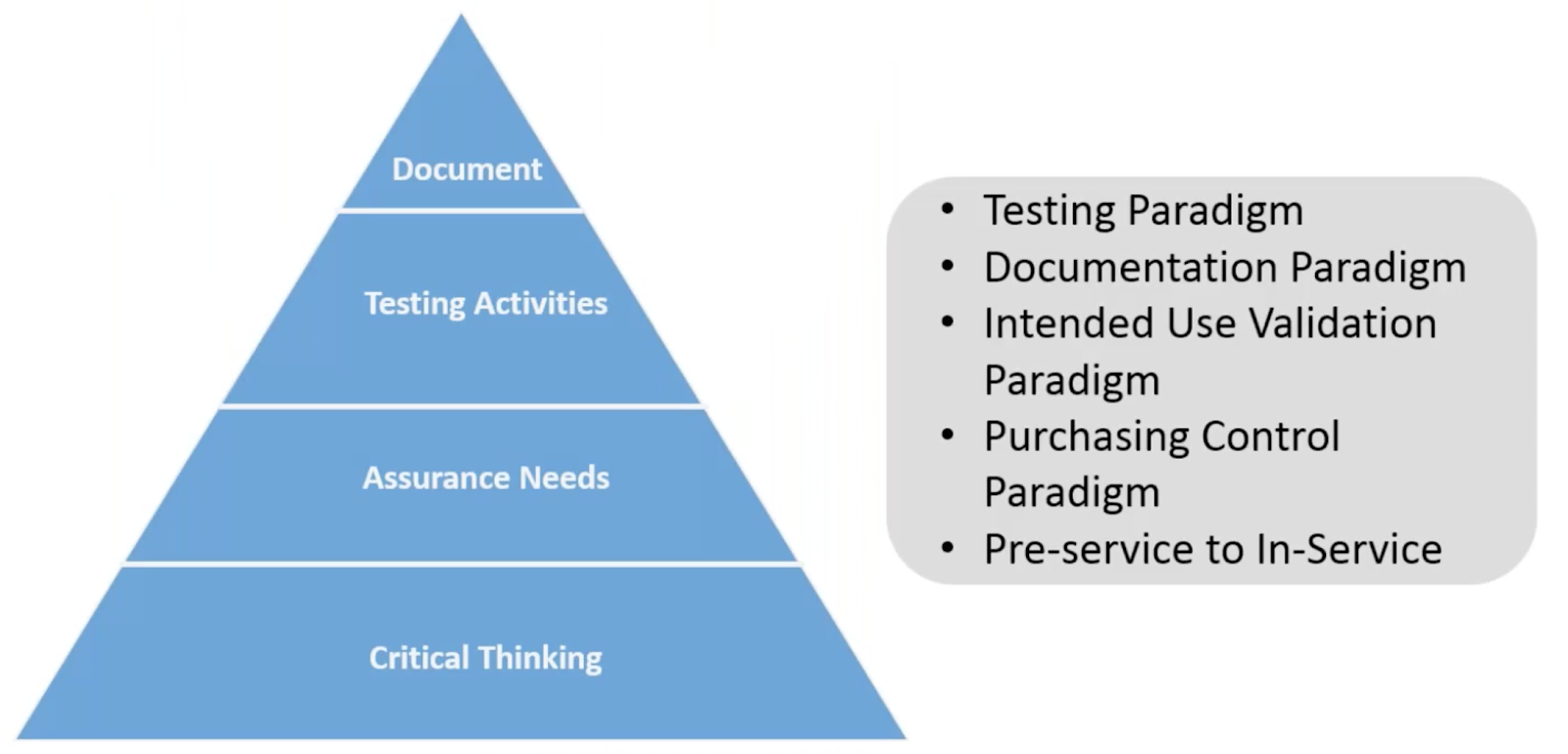
The image above illustrates the current paradigm regarding typical CSV efforts. The current paradigm is skewed towards heavily documenting validation protocols and testing results with little emphasis on assurance needs and critical thinking.
You see, all too often we choose the documentation path as proof that validation needs have been addressed. We draft specific test cases and protocols and riddle our test results with screenshots as objective evidence to demonstrate certain criteria have been met.
Yes, there is value in documenting decision-making process. And yes, the current approaches we have used are more of a compliance, check-box way of demonstrating software has been validated.
Flip the CSV Script
FDA would like to encourage you to “flip the script” on CSV.
Source: Cisco Vicenty, FDA Case for Quality Program Manager
Documentation should be the least burdensome activities for CSV while critical thinking should be bulk of focus for CSV efforts. This requires shifting from “validation” towards more of “assurance” in terms of mindset.
And in order to make this shift consider the following questions:
- How can you demonstrate confidence system is meeting your needs and intended use?
- What goals are you looking to achieve?
I can imagine what you are thinking. You have been conducting CSV with a document centric focus and compliance-based approach for a long time. How in the world are you going to flip your script and apply more critical thinking and focus on software assurance? (And is FDA really going to shift its focus this way too?)
Critical Thinking & Software Assurance
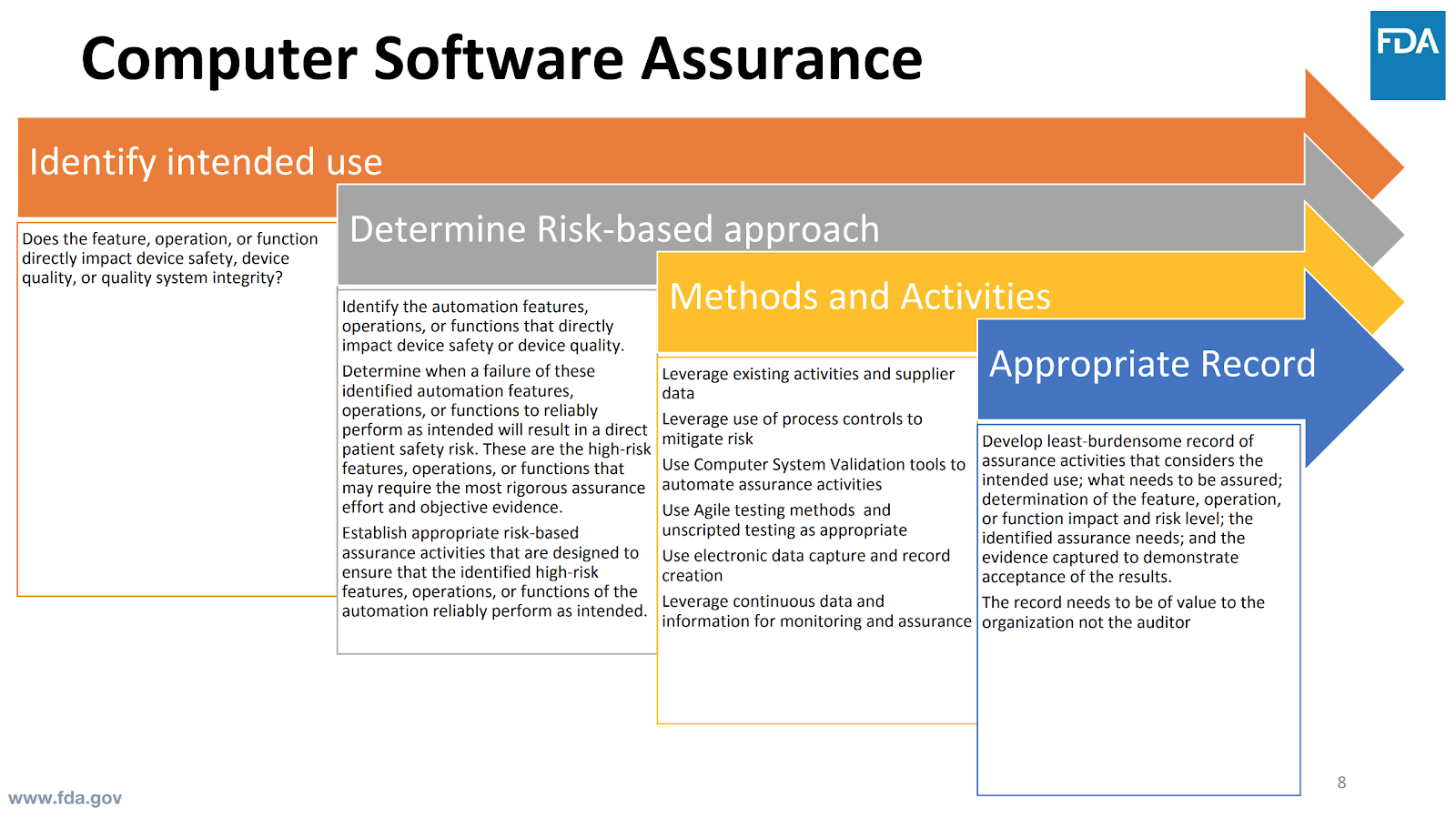
In order to make this shift, start with understanding your intended use of the system.
To do so, ask yourself these questions:
- Does this impact patient safety?
- Does this impact device quality?
- How does this influence your quality system integrity?
Source: Cisco Vicenty, FDA Case for Quality Program Manager
Then begin to apply critical thinking by using a risk-based approach to assess the system features, focusing on direct impact to patient safety and device quality. By applying risk-based methodology, you will need to assess what happens in the event there is a failure of the system.
From there, you can start to define the type and level of assurance activities you will need to ensure the features, operations, and function perform reliably.
Keep in mind that even current regulations do not prevent you from applying risk-based approaches for assurance activities. However, interpretation has been vague, at best. Focus on patient safety. Does failure cause a harm situation? This drives the rigor of assurance activities.
What are the appropriate records to demonstrate assurance? Define what is the least-burdensome assurance record(s) required. When possible, leverage existing supplier data, information, and testing records.
You get to define what is appropriate based on the identified risks of the system. The records need to provide value to you--not just to satisfy the auditor.
Let me make a note about quality management system integrity and relationship with CSV and this new paradigm. Yes, QMS integrity is important and largely part of demonstrating compliance.
FDA views this as important, yet low risk with respect to patient safety risk. Why does this matter? By applying risk-based critical thinking to your QMS automation efforts has potential to reduce your validation burden.
Note that when leveraging supplier records, even those from your eQMS provider, it is important for you to apply critical thinking and a thorough review of these records. Do not just take the supplier records and check a box. You need to ensure that the records provided are appropriate and address your intended uses of the systems and risks identified.
This is a huge part of what makes Greenlight Guru different from other Quality Management Software options. Greenlight Guru is purpose-built specifically and exclusively for the medical device industry by experienced medical device professionals. Our focus on enabling companies to reach true quality is coupled with our commitment to advancing industry best practices through our partnership with the FDA on their Case for Quality.
Significant regulatory burdens are removed for manufacturers by ensuring all documentation and records pertaining to eQMS validation are in alignment with regulations and current FDA expectations.
Understanding Software Assurance
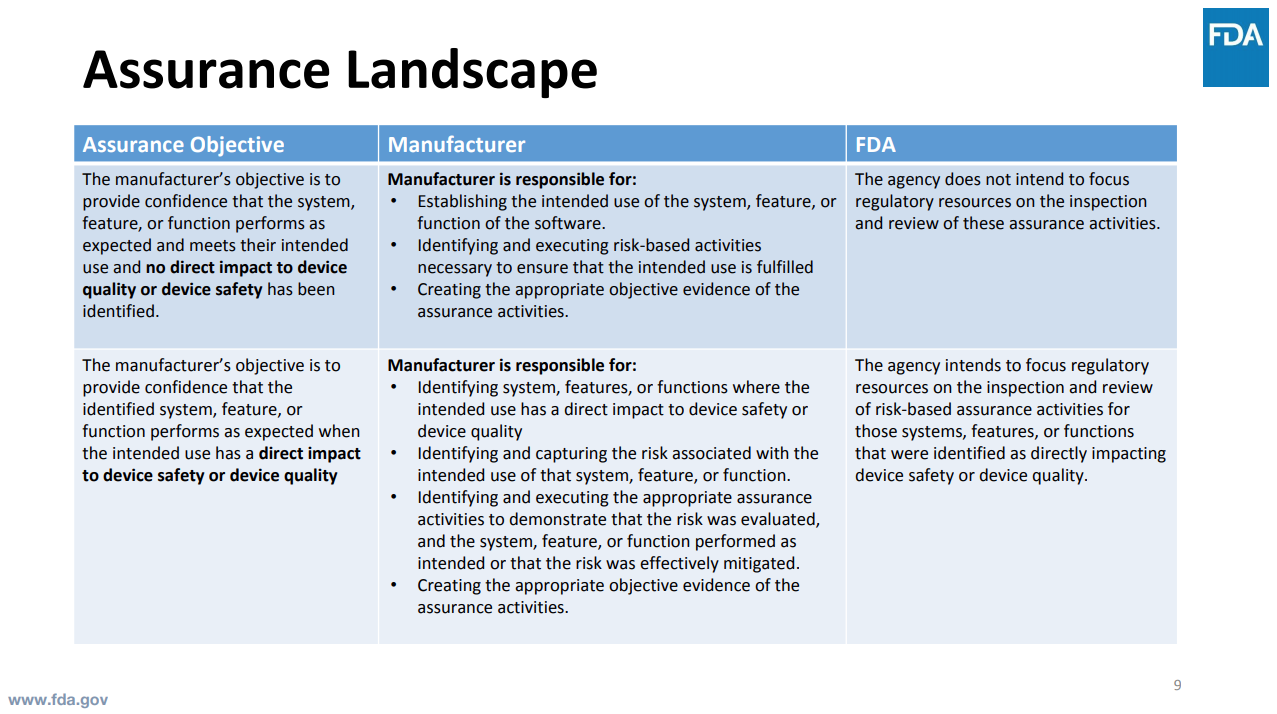
Software assurance is about you demonstrating and having confidence that the system meets your intended use and the features and functions perform as expected without impacting patient safety and device quality.
Source: Cisco Vicenty, FDA Case for Quality Program Manager
FDA does not want to direct resources to inspect and review assurance activities. Instead, FDA expects the medical device company to be responsible for doing so.
What does FDA care about? FDA is going to focus time and effort on direct impact to device quality, device safety, direct patient safety risks.
Specifically:
- Directly impacts physical properties of the product or manufacturing process identified as essential to device safety or device quality by the manufacturer
- Measures, inspects, analyzes, and or dispositions the product or process
- Determines acceptability or performs process corrections without human intervention, awareness, or review
- Directly impacts labeling, instructions for use, or direct alerts or communications to the user
- Automates surveillance, trending, or tracking of product quality or patient safety issues identified as essential by the manufacturer
And this does not mean that if the areas listed above are impacted that FDA expects you to necessarily address via robust protocols, testing, etc. This does mean FDA cares about these areas and expects you to focus on critical thinking and assurance.
Remember, you do not need to reinvent the wheel. Leverage existing activities. Leverage supplier data and information. Use CSV tools to automate assurance activities.
Note: FDA is not interested in spending resources to inspect validation of these CSV tools. You will determine the correct tools and assurance activities.
When applicable, use agile testing methodologies. Use electronic data capture and record creation. “Paper” documentation and/or screenshots are not always necessary and often provide little to no actual value.
Identify the least burdensome records that address software assurance.
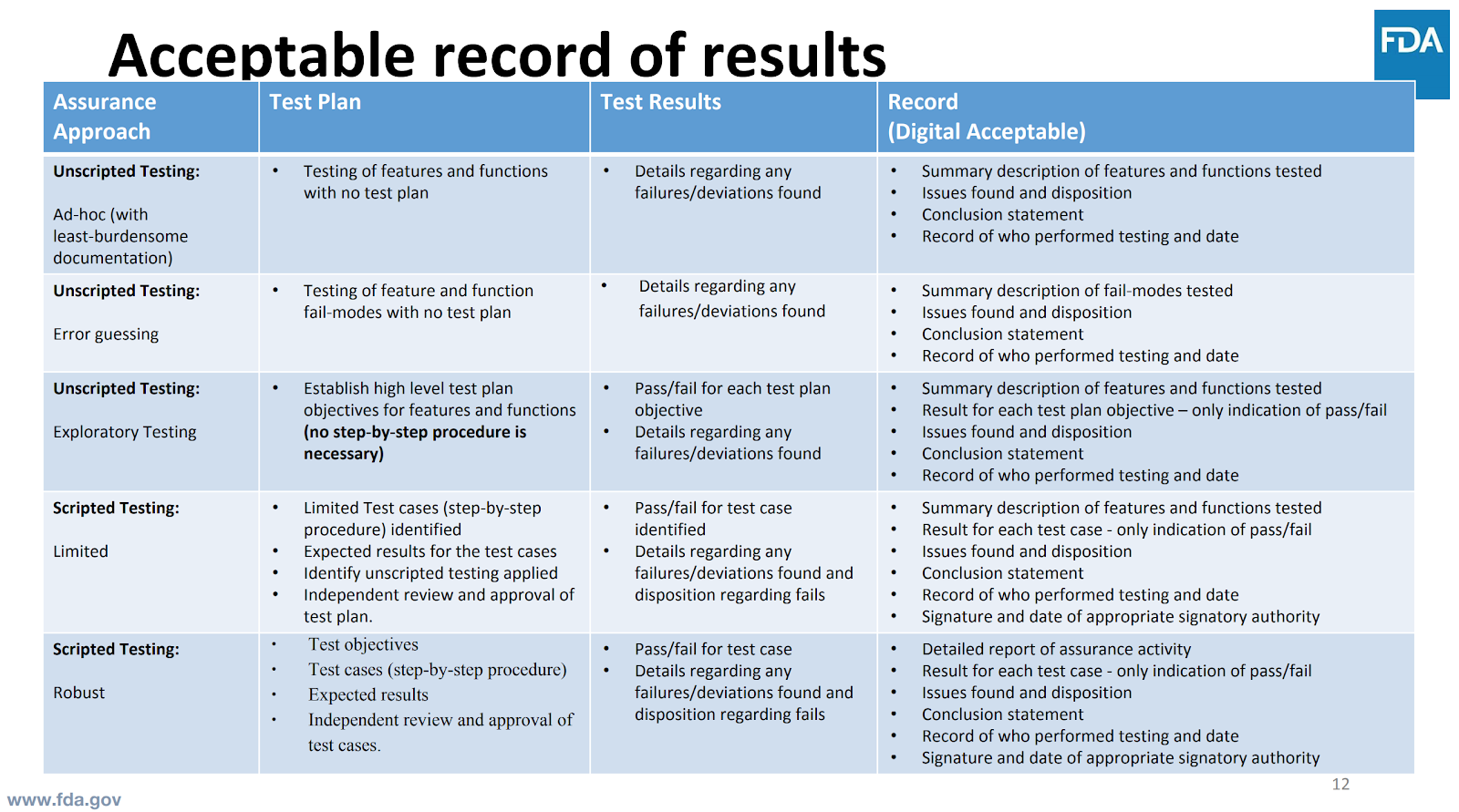
Source: Cisco Vicenty, FDA Case for Quality Program Manager
Start your shift toward software assurance today
You don’t have to wait for this guidance to shift your mindset towards true quality when evaluating opportunities to embrace automation and technology.
You can apply this new way of thinking and approaches today. Doing so will help you with better information, product performance knowledge, easier methods to track and trend, more timely responsiveness, process optimization, reducing in product risks, reducing operational expenses, and increasing business value.
Ultimately, embracing automation and technologies within your business has the great potential to improve your product quality and reduce patient risks.
Looking for an all-in-one QMS solution to advance the success of your in-market devices and integrates your quality processes with product development efforts? Click here to take a quick tour of Greenlight Guru's Medical Device QMS software →
FDA Case for Quality: 2018 Comprehensive Review





