
With the deadline for compliance with the EU MDR just around the corner, big changes are being made by medical device companies selling in the European Union marketplace. The new EU Medical Device Regulation (MDR (EU) 2017/745) is replacing the Medical Device Directive (MDD) and the Active Implantable Medical Devices Directive (AIMDD).
The new MDR aims to improve product quality, reliability, safety, and performance. Several of the new requirements involve an increased focus on traceability and transparency throughout the medical device supply chain, and more clinical evidence is now required to support this.
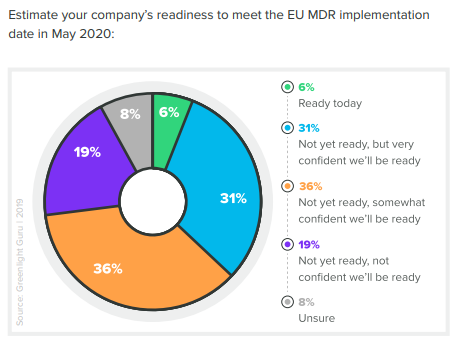
All in all, the EU MDR should make for a healthier medical device landscape in the EU once implemented. However, our 2020 State of Medical Device Product Development and Quality Management Report showed that as of the end of 2019, only 6% of companies felt prepared for the EU MDR transition.
Manufacturers know that rushing to achieve compliance greatly increases the chances of human error. Mistakes made during this transition process can impede progress and cause immediate and long-term issues.
Such mistakes can add costly delays to companies bringing a medical device to market or result in quality issues, complaints, and recalls for devices already on the market. However, there’s still time to carefully prepare your transition to the EU MDR.
We’ve prepared a road map to help you prepare for the EU MDR. Let’s start by taking a look at the biggest changes under the new regulation and then we'll provide three easy steps you can take to complete your transition for compliance.
Biggest Changes Under EU MDR
Many medical device processes have new requirements under the new medical device regulation. Significant changes to post-market processes including post-market surveillance, traceability, labeling, and clinical evidence are among the top changes impacting manufacturers.
As stated by the European Commission, manufacturers must establish, document and implement a quality system, and maintain its effectiveness throughout the total device life cycle.
Total Life Cycle Traceability
The EU MDR requires total life cycle traceability between all stages of product development and post-market activities. This is where many of the companies we surveyed feel underprepared.
According to our report findings, only one in four companies is “very confident” they could demonstrate total life cycle traceability in the face of an unannounced audit.
When medical device manufacturers were asked which device standards, principles, or regulations they found to be most difficult to implement, the most commonly cited were EU Medical Device Regulation (38%), Risk Management Standard — ISO 14971 (23%), and Computer System Validation (22%).
Implementing a quality system that provides total life cycle traceability will automate the ability to connect post-market quality data back into design activities. This type of purpose-built QMS solution allows teams to focus on value-added activities and eliminates cumbersome paper-pushing, often found with legacy and paper-based systems, that is highly error-prone and inefficient.
Post-market Surveillance Reporting (PMSR)
Under the European medical device regulation, companies with low-risk Class I devices are required to produce a post-market surveillance report. Your PMSR must summarize the results and conclusions of PMS data, along with a rationale and description of any corrective actions taken for devices on the market. This report will then become part of your technical documentation.
As outlined in Chapter VII Section 2 of the medical device regulation, new requirements state that post-market surveillance reporting must be handled within 15 days at maximum, with three different time frames:
-
Reporting a serious threat to public health must be carried out in 2 days.
-
Reporting a death or serious health deterioration must be done within 10 days.
-
Reporting all other serious incidents must be done within 15 days.
You should know that the new regulation also places a strong emphasis on continuously updating the risk, technical file and ongoing evaluation of your devices.
Companies will need to make sure their quality system is built with the infrastructure to capture, process, and report these incidents within these new time frames. As with the requirements for total life cycle traceability, post-market surveillance reporting will require a robust QMS that can connect key processes throughout the product life cycle with efficiency and ease.
Labeling
Medical device labeling, in particular, has been overhauled in the new EU MDR. Additional requirements have been added, such as the need to provide an general safety and performance requirements (GSPR) checklist. You now must provide information on warnings, precautions or contraindications on your device labeling. For class III and implantable devices, a summary of safety and clinical performance is required on the labeling.
Labels now require the EC representative’s name, address, and symbol. The EU MDR also dictates that a new symbol be included on the label to identify its product as a medical device. Device labeling will also need to be published electronically on the manufacturer’s website as well as in print, and manufacturers are required to publish user data on their websites, as well.
Once EUDAMED is live in May 2022, you will need to establish a procedure for uploading your Unique Device Identification (UDI) data to the database. This applies to all medical devices.
Clinical Evidence
EU MDR mandates that clinical evidence is now required for all medical devices. This new requirement will involve extensive clinical testing in some cases, causing some companies to panic, namely those with older devices that do not have this evidence readily available.
Such testing will be costly and time-consuming, so companies should plan accordingly. Manufacturers with diverse portfolios of devices will bear the largest burden when it comes to new clinical testing.
Unfortunately for some companies, undergoing this process will not be financially feasible from a cost/benefit perspective. Some devices may be low-sale products, and the cost of clinical testing could outweigh the benefit of retaining their product on the market.
In these cases, companies will be faced with the decision of whether to cut certain products or choose new markets that are more cost-effective to sell the device.
With the EU MDR placing emphasis on total life cycle traceability, it's important to note this includes the entire supply chain, which the regulation refers to as Economic Operators. This means any third-party suppliers and manufacturers involved at any stage of your product's life cycle will also need to be evaluated for compliance with these requirements.
3 Ways to Transition for EU MDR Compliance
Preparing your medical device company for the EU MDR is understandably a major undertaking. The good news is, there is still time to make the transition. Once you’ve made a plan, you’ll find it far easier to stay on target and achieve compliance before the 26 May 2020 deadline.
1. Review The New EU Medical Device Regulation
The first step you can take is to familiarize yourself with the detailed requirements of the Regulation (EU) 2017/745. While we’ve included the new changes that require your attention as you prepare, the regulation is a 566-page document, so the sooner you can start the better.
It’s important that you take the time to go through each section in detail or identify a quality and regulatory subject-matter expert who can be entrusted with this task. They’ll help you understand the specific requirements and ensure you're in compliance with them.
2. Perform A Gap Assessment
After you’ve familiarized yourself with the new requirements, you should conduct a gap assessment of your existing quality system processes in order to understand, assess, and execute the necessary changes for compliance. We published a free MDD vs. MDR gap assessment tool to help you get started.
A gap assessment is a valuable tool that will help you identify which requirements apply to your specific medical device, while ruling out the ones that do not. Upon completion, you should have a clear understanding of your to-do's for fully transitioning to the new regulation.
Keep in mind that the results of your gap assessment may show significant changes that are too much for one, single person to manage. Breaking the entire process down into multiple teams and stages will make your journey to compliance faster and more manageable. You can do this by delegating responsibility to manage the changes as sub-projects.
3. Conduct Internal Audits
This stage of the process will be one of the most time-consuming for most companies, since there’s a lot to consider with the new regulations. Here’s a list of items you must assess during your internal audit:
- General Safety and Performance Requirements
- Technical File
- Risk Management activities, in accordance with ISO 14971, that align with PMS and PMCF activities
- Unique Device Identification (UDI) system
- Post-market Surveillance (PMS) and Post-market Clinical Follow-up (PMCF)
- Clinical Evaluation
- Labeling
- EUDAMED (once available) process
- Common Specifications
The Audit Management capabilities of Greenlight Guru's QMS software give companies full visibility into all quality management processes. Our audit workflow allows you to create schedules, specify due dates and reminders, assign owners, and easily share with others, and even connects audit findings with quality events that may be linked.
As we've covered at length in this article, total life cycle traceability and connectivity are key factors for compliance with MDR.
Is Your QMS Ready for EU MDR?
One key area in which companies could experience issues with EU MDR compliance is through the use of inefficient quality systems. The regulation requires that systems and documentation maintain compliance throughout the entire life cycle of a device. This means every single system and process must all be accounted for, stored, and readily accessible within a company’s QMS.
It’s imperative that your Notified Body is able to easily find everything they need to prove full traceability and compliance during an audit. This can prove challenging, depending on the type of QMS you have in place.
With legacy systems, documents can easily get lost or accidentally duplicated when stored using paper-based or general-purpose tools, like Google Drive or Dropbox. This document storage problem is only exacerbated when using or incorporating physical paper documents.

A general-purpose QMS is just that – general purpose. These solutions lack the purpose-built capabilities that medical device companies need to integrate their core QMS processes to achieve total life cycle traceability for compliance with MDR. Not to mention, the configuration, implementation, and management of these quality systems will cost you more time and money in the long run.
We recommend considering multiple options when choosing the best QMS to suit your needs. A purpose-built QMS solution for medical devices should have clear audit trails and real-time workflows that require little to no configuration for immediate use, including out-of-the-box alignment with industry best practices from EU MDR, ISO 14971, and ISO 13485.
Making the Full Transition to EU MDR
There’s a lot to consider, but by familiarizing yourself with the new regulation, performing a gap assessment, conducting internal audits, and evaluating your QMS, your full transition is possible. In fact, doing this will place your company ahead of 94% of other device makers that are unprepared for EU MDR to go into effect in May 2020.
At Greenlight Guru, we create these resources because we’re passionate about guiding companies toward success in producing safe, effective medical devices. That’s also why we created the only QMS Software purpose-built for medical devices on the market.
Our purpose-built QMS software solution is designed as a a closed-loop quality system that facilitates total life cycle traceability for medical device companies. If you're looking for an all-in-one QMS solution to advance the success of your in-market devices that also integrates your post-market activities with product development efforts, click here to take a quick tour of Greenlight Guru's QMS software.
Free Download: MDD vs. MDR Gap Assessment Tool






