

















Table of Contents
Compliance isn’t a pop quiz. The teacher—the U.S. Food and Drug Administration (FDA), in this case—has given you the answers to the test. Now, we’re not saying this open note test makes regulations like 21 CFR Part 820 easy to follow. But the challenge is different than you might first assume. What separates market leading medical device companies from the rest isn’t knowledge—it’s implementation.
Much, if not all, of the information is out there. The medical device companies that excel are the ones that can take it all in and execute—both in terms of understanding the big picture (What is 21 CFR Part 820? What does it cover? What is its purpose?) and the granular details (What goes into an approved supplier list?).
This in-depth, easy to understand guide paints a comprehensive picture of 21 CFR Part 820 by explaining the regulations in an easy to understand way. In addition to answering the aforementioned questions throughout this guide, each subpart of the regulation is accompanied by specific advice on how you can comply as well as common mistakes manufacturers face so you can make sure to avoid the pitfalls.
Let's get started by reviewing the basics of 21 CFR Part 820 before diving into the details of the regulatory requirements that you must follow in order to comply with FDA’s quality system regulation for medical devices.
FREE RESOURCE: Click here to download the eBook of our Ultimate Guide to 21 CFR Part 820.
 What is 21 CFR Part 820?
What is 21 CFR Part 820?
21 CFR Part 820 is a set of regulations from FDA that outlines the current good manufacturing practice (CGMP) requirements that medical device manufacturers in the United States must follow with regards to their quality system. These CGMP requirements ensure medical device companies establish a QMS that enables the delivery of safe, effective, and compliant products.
As stated by FDA, 21 CFR Part 820 covers “the design, manufacture, packaging, labeling, storage, installation, and servicing of all finished devices intended for human use,” including the facilities and designs used for those processes.
Structure of the quality system regulation for Medical Devices
21 CFR 820, though a dense document, lays out medical device quality system regulations (QSR) in a digestible way for manufacturers to best interpret and apply to their own specific device. The QSR consists of 15 subparts and is structured by way of order from big picture questions regarding scope to detailed rules about what manufacturers should do and when.
The very first section, Sec. 820.1 Scope, lays out the applicability, authority, and exemptions involved. It explains the intention of 21 CFR Part 820 (“to ensure that finished devices will be safe and effective”) and to whom it applies (“manufacturers of finished medical devices”). Important to this latter point, it notes that manufacturers that only engage in parts of the medical device manufacturing process only need to comply with relevant requirements.
This first section also establishes a rule that applies throughout the regulation: if a conflict between regulations emerges, “the regulation specifically applicable to the device in question shall supersede the more general.”
Section 820.3 provides definitions of 30 different terms used throughout 21 CFR Part 80.

These definitions, the first five of which are shown above, ensure manufacturers and regulators share a common understanding of the terminology in use. If you’re confused about the meaning of a term, refer to Section 820.3 or lean on trusted, outside resources, such as the Greenlight Guru list of the top medical device terminology.
What is the difference between the FDA QSR and ISO 13485?
21 CFR Part 820 has a close relationship with ISO 13485:2016, especially after the 2016 revision of ISO 13485, which closely aligns to the requirements found in FDA’s QSR. In February of 2022, FDA released its proposed rule to harmonize its QSR with ISO 13485:2016 for the new Quality Management System Regulation (QMSR).
The main difference between 21 CFR 820 and ISO 13485:2016 is that ISO 13485 is a voluntary standard that defines quality system requirements. 21 CFR Part 820 comes from FDA, a federal agency, meaning that noncompliance can incur punitive measures that range from citations to recalls to fines and, in rare cases, to litigation. While FDA is local to the United States, much of the world recognizes and follows ISO standards.
What are the FDA Requirements of 21 CFR Part 820—Quality System Regulation?
FDA conducts regular inspections to ensure compliance with 21 CFR 820. FDA uses the Quality System Inspection Technique (QSIT) to evaluate the alignment of internal quality system processes with regulatory requirements. The stakes of compliance are clear. Violations can result in 483 observations and warning letters.

21 CFR Part 820 Subpart B—Quality System Requirements
What it covers: 21 CFR Part 820 Subpart B covers management responsibility, internal quality audits, and personnel. This includes a range of roles, as well as what each role is in charge of doing and what training they require.
How to comply: §820.20, the first section of 21 CFR Part 820 Subpart B, covers management responsibility that includes regulations about policies, resources, and planning:
-
Quality policy: Commit to quality and ensure the company understands quality, as well as implements and maintains it.
-
Organization: Use an organizational structure commensurate with the production of compliant medical devices, including handing out appropriate responsibilities to appropriate personnel and providing the resources necessary to meet FDA requirements.
-
Management review: Review the QMS at regular intervals and document those reviews.
-
Quality planning: Plan out the practices, resources, and activities that support quality.
-
Quality system procedures: Set quality system procedures and instructions.
820.25, the third section of 21 CFR Part 820 Subpart B, covers Personnel. This section, as §820.20 mentions, lays out the requirements for manufacturers to have personnel who are experienced enough to ensure compliance. That includes providing training that covers potential defects these personnel might witness as a result of noncompliant activities.
Common mistake: Internal quality audits are often an area where manufacturers struggle. Many companies treat internal quality audits as a checkbox activity rather than a value-added activity, meaning they work toward mere compliance instead of quality outcomes.
There are two ways to make the shift from compliance-only to true quality:
-
Establish a quality culture that treats quality as an asset worth investing in.
-
Right-size your QMS so that the size and complexity of your quality system procedures are actually simple enough for a company of your current size to manage.
The key to conducting successful internal quality audits is to take a proactive instead of a reactive approach.
![21 CFR Part 820 Design Controls]() 21 CFR Part 820 Subpart C—Design controls
21 CFR Part 820 Subpart C—Design controls
 21 CFR Part 820 Subpart C—Design controls
21 CFR Part 820 Subpart C—Design controlsWhat it covers: 21 CFR Part 820 Subpart C covers design controls. Design controls are procedures that ensure devices are designed according to their requirements.
How to comply: 21 CFR Part 820 Subpart C first lays out which devices this section applies to. This includes Class I devices, such as devices automated with computer software, as well as the devices in the chart below.

21 CFR Part 820 Subpart C includes additional requirements during the design process:
-
Design and development planning: Have regularly reviewed plans for design and development.
-
Design input: Ensure design requirements fit the intended use of the device, including a way for manufacturers to address “incomplete, ambiguous, or conflicting requirements.”
-
Design output: Define and document design outputs in such terms that you can evaluate conformance to design inputs.
-
Design review: Plan and conduct regular design reviews that include people involved with the reviewed function, people uninvolved, and specialists.
-
Design verification: Check that design outputs match design inputs.
-
Design validation: Check that devices, including software, match defined user needs and intended uses. This can involve testing production units.
-
Design transfer: Translate your device into production specifications.
-
Design changes: Review design changes before you implement them.
-
Design history file (DHF): Create a DHF with records that show you designed a compliant device according to your plans.
Common mistake: Some companies wait too long to establish design controls, which can result in extra time and rework. The key is to establish your design controls early, so you can reliably capture all the data you need.
Design controls are what we call a systematic framework. Your company identifies and tracks which parts of the medical device manufacturing process prove your product meets user needs, demonstrates safety, and confirms effectiveness.
While some aspects of your QMS can be delayed, design controls cannot.
![21 CFR Part 820 Document Controls]() 21 CFR Part 820 Subpart D—Document controls
21 CFR Part 820 Subpart D—Document controls
 21 CFR Part 820 Subpart D—Document controls
21 CFR Part 820 Subpart D—Document controlsWhat it covers: 21 CFR Part 820 Subpart D covers document controls. Document control refers to policies and procedures quality managers use to manage documents throughout the medical device product lifecycle.
How to comply: 21 CFR Part 820 Subpart D lays out regulations for document approval and distribution as well as document changes:
-
General: Establish procedures that control documents so that they comply with 21 CFR Part 820.
-
Document approval and distribution: Designate one person or a group of people to review and approve documents for their compliance.
-
Document changes: Review document changes in the same fashion you approve and distribute them.
Common mistake: Many companies err on the side of caution and over-document to a laborious extent. Instead, document what you need to demonstrate compliance. When documents become overly complex, they become unmanageable. When documents become unmanageable, compliance becomes difficult.
That said, too little documentation can expose a disconnect between the applicable regulations and your finished device. The best advice is to keep your document control simple. Be proactive and don’t wait to do documentation after the product is released.
![21 CFR Part 820 Purchasing Controls]() 21 CFR Part 820 Subpart E—Purchasing Controls
21 CFR Part 820 Subpart E—Purchasing Controls
 21 CFR Part 820 Subpart E—Purchasing Controls
21 CFR Part 820 Subpart E—Purchasing ControlsWhat it covers: 21 CFR Part 820 Subpart E covers purchasing controls. This includes the “evaluation of suppliers, contractors, and consultants,” as well as purchasing data.
How to comply: 21 CFR Part 820 Subpart E requires you to ensure compliance of the products and services you receive, specifically:
-
Evaluation of suppliers, contractors, and consultants: Set quality requirements for your suppliers, contractors, and consultants. Based on those requirements, evaluate and select said third parties, and define the control you need over what the third parties supply.
-
Purchasing data: Track the quality requirements, including agreements between the manufacturer and third party about changes in what the third party will supply.
Common mistake: Many medical device companies aren’t as careful as they could be when selecting suppliers. 21 CFR Part 820 suggests manufacturers set out qualification requirements, but does not specify how stringent these should be, leaving many medical device companies with mediocre supplier management processes.
Truly effective supplier management means conducting due diligence audits of your suppliers, which can be done by sending out a supplier survey or by keeping an approved supplier list (ASL) to track which suppliers you’ve vetted and approved.
![21 CFR Part 820 Identification and Traceability]() 21 CFR Part 820 Subpart F—Identification and Traceability
21 CFR Part 820 Subpart F—Identification and Traceability
 21 CFR Part 820 Subpart F—Identification and Traceability
21 CFR Part 820 Subpart F—Identification and TraceabilityWhat it covers: 21 CFR Part 820 Subpart F covers identification and traceability. Identification refers to the ability to avoid product “mixups,” and traceability refers to the ability to trace product defects back to their source.
How to comply: 21 CFR Part 820 Subpart F includes only two short sections, the first of which is a single sentence long:
-
Identification: Be able to identify products as you receive, produce, distribute, and install them.
-
Traceability: Be able to identify the units, lots, and batches of finished devices that can cause significant injury when they fail. Establish procedures for corrective action as a result and document traceability in your DHR.
Common mistake: Original data from the 2020 State of Medical Device Report shows that traceability is hard to establish. Closed-loop traceability—when companies can connect post-market quality data and design activities—is rare among medical device companies.
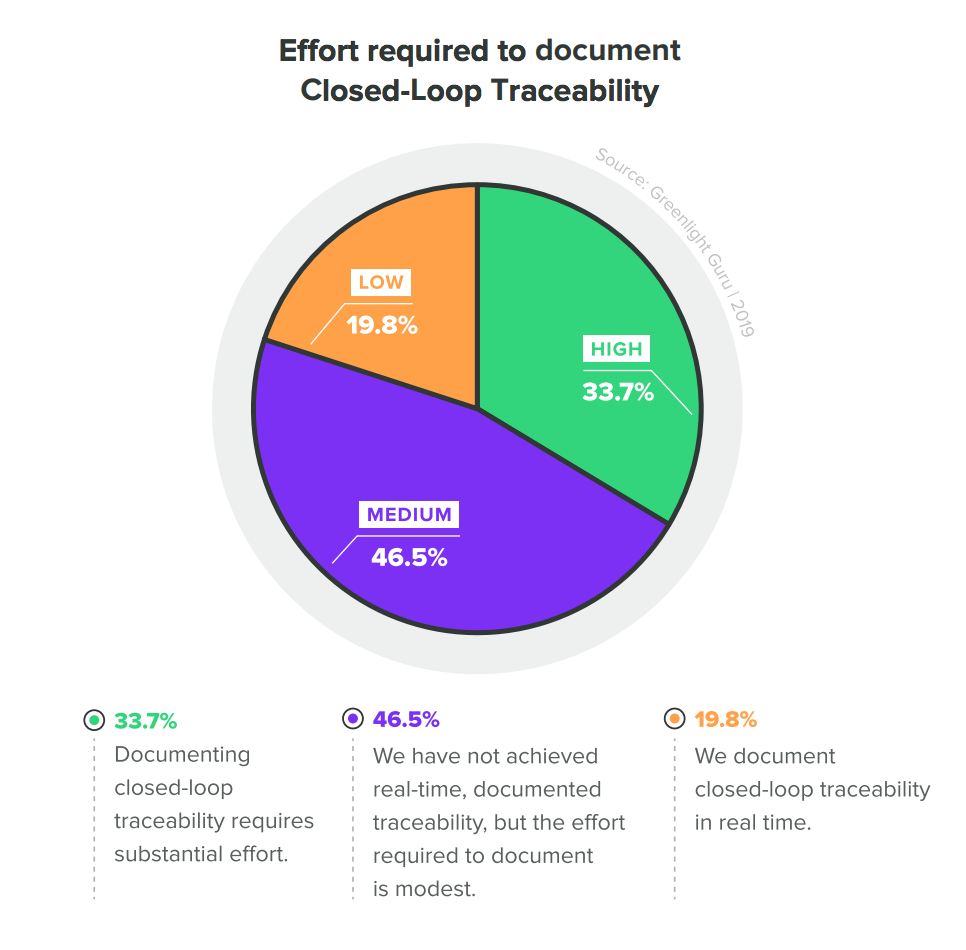
Eight out of ten medical device companies struggle with documenting closed-loop traceability (CLT) in real time. The key is to leverage the best QMS software, a solution that replaces inefficient legacy systems that are inherently not capable of achieving CLT.

As the graph above shows, legacy tools make medical device companies significantly less confident in demonstrating traceability. Companies that invest in a modernized solution, like Greenlight Guru, can achieve closed-loop quality system traceability with ease. See your quality system like never before by getting a free demo of Greenlight Guru today.
![21 CFR Part 820 Production and Process Controls]() 21 CFR Part 820 Subpart G—Production and Process Controls
21 CFR Part 820 Subpart G—Production and Process Controls
 21 CFR Part 820 Subpart G—Production and Process Controls
21 CFR Part 820 Subpart G—Production and Process ControlsWhat it covers: 21 CFR Part 820 Subpart G covers Production and Process Controls. Controls include the inspection, measuring, and test equipment, as well as process validation.
How to comply: 21 CFR Part 820 Subpart G explains how you can control the device production process.
-
General: Develop and monitor production processes that manufacture devices according to your specifications. Document controls necessary to ensure conformance, including instructions and standard operating procedures (SOPs).
-
Production and process changes: Create procedures for making, verifying, and documenting changes to specifications, methods, processes, or procedures.
-
Environmental control: Control any relevant environmental conditions that might affect product quality.
-
Personnel: Ensure contact between staff and product doesn’t damage product quality.
-
Contamination control: Prevent your equipment or products from getting contaminated.
-
Buildings: Use buildings that can sufficiently house the manufacturing process.
-
Equipment: Check that all manufacturing equipment meets requirements. Set a schedule for maintenance as well as periodic inspections.
-
Manufacturing material: Use, discard, and document manufacturing material that could damage product quality.
-
Automated processes: Validate computer software, including any software changes, for its intended use.
Subsection §820.72 hones in on details regarding the control of inspection, measuring, and test equipment:
-
Control of inspection, measuring, and test equipment: Check that your inspection, measuring, and test equipment matches their intended purposes and can produce valid results.
-
Calibration: Include accuracy and precision directions for calibration as well as guides on what to do if directions aren’t followed. Trace calibration standards to national or international standards. Document every calibration.
Subsection §820.75 focuses on process validation:
-
Validate processes that you can’t fully verify with inspections and tests.
-
Monitor and control validated processes so that you can keep meeting requirements. Qualified people should perform the validations and document them.
-
Re-validate as necessary when changes or deviations happen.
Common mistake: Process validation is one of the most common areas prone to mistakes by companies when implementing 21 CFR Part 820. These requirements are found to be most challenging for companies that try to validate their processes from scratch and, in so doing, struggle to correctly interpret the regulation.
The goal of process validation is to demonstrate that you can replicate your processes. Each time you run a validated process, you expect the same level of quality every time.
Ensuring this level of replicability is hard to do from scratch using ad hoc systems. With a modern QMS from Greenlight Guru, however, you can use pre-validated components. These components include templates and workflows that have weathered a multitude of audits.
![21 CFR Part 820 Acceptance Activities]() 21 CFR Part 820 Subpart H—Acceptance Activities
21 CFR Part 820 Subpart H—Acceptance Activities
 21 CFR Part 820 Subpart H—Acceptance Activities
21 CFR Part 820 Subpart H—Acceptance ActivitiesWhat it covers: 21 CFR Part 820 Subpart H covers acceptance activities. This includes receiving, in-process, and finished device acceptance as well as regulations for acceptance status.
How to comply: 21 CFR Part 820 Subpart H explains how you can properly accept incoming products:
-
General: Establish procedures for inspections, tests, or other types of verification—all known as acceptance activities.
-
Receiving acceptance activities: Inspect, test, or verify that incoming product matches requirements.
-
In-process acceptance activities: Check that in-process product also meets requirements.
-
Final acceptance activities: Verify that production runs, lots, or batches of finished medical devices fulfill acceptance criteria. Control the devices until you’re ready to release them.
-
Acceptance records: Document required acceptance activities.
Subsection §820.86 digs into the definition of acceptance status. Acceptance status indicates whether a product conforms with pre-established criteria. FDA emphasizes the ability of the manufacturer to identify acceptance “throughout manufacturing, packaging, labeling, installation, and servicing of the product.”
Common mistake: If your inspection doesn’t have a structure to it, it will be difficult to ensure its effectiveness. Including criteria, so you’re not inspecting ad hoc, will help manufacturers avoid this common mistake. You always need to confirm that your specifications and acceptance criteria have been met.
![21 CFR Part 820 Nonconforming Products]() 21 CFR Part 820 Subpart I—Nonconforming Product
21 CFR Part 820 Subpart I—Nonconforming Product
 21 CFR Part 820 Subpart I—Nonconforming Product
21 CFR Part 820 Subpart I—Nonconforming ProductWhat it covers: 21 CFR Part 820 Subpart I covers nonconforming product. This includes control of nonconforming product as well as review, disposition, and rework.
How to comply: 21 CFR Part 820 Subpart I requires manufacturers to keep close control over products that don’t conform to requirements:
-
Control of nonconforming product: Control product that doesn’t conform to requirements, including “identification, documentation, evaluation, segregation, and disposition.” Determine whether an investigation is necessary.
-
Nonconformity review and disposition: Determine who’s responsible for disposing of nonconforming product and the documentation procedures for why and how. Additionally, prepare procedures for rework and reevaluation of previously nonconforming product.
Common mistake: A recurring compliance issue that plagues manufacturers with 483 notifications are inadequate procedures for controlling nonconforming product. Make sure you can identify nonconforming product and keep it segregated from conforming product.
After dealing with nonconforming product, companies often forget to document the nonconformance process correctly. Download this free nonconformance template to help you streamline the reporting process. With the right documentation, you can better assess when you’ll need to investigate more deeply.
![21 CFR Part 820 CAPA]() 21 CFR Part 820 Subpart J—Corrective and Preventive Action (CAPA)
21 CFR Part 820 Subpart J—Corrective and Preventive Action (CAPA)
 21 CFR Part 820 Subpart J—Corrective and Preventive Action (CAPA)
21 CFR Part 820 Subpart J—Corrective and Preventive Action (CAPA)What it covers: 21 CFR Part 820 Subpart J covers corrective and preventive action, better known as CAPA. CAPA is the process whereby medical device companies address systemic flaws in the medical device manufacturing process.
How to comply: According to FDA, compliant CAPA procedures must:
-
Identify causes of nonconforming product among “processes, work operations, concessions, quality audit reports, quality records, service records, complaints, returned product, and other sources of quality data.”
-
Investigate the causes of nonconformance.
-
Determine what you need to do to correct and prevent the nonconformance from recurring.
-
Verify and validate your CAPA.
-
Implement and record necessary changes.
-
Make issues about quality problems known to people with relevant roles.
-
Submit CAPA and quality problems to management.
-
Document everything involved in identifying and resolving your CAPA.
Common mistake: Not all product defects should cause CAPA. Isolated incidents can be dealt with through minor corrections. You can ensure you’re following best practices for CAPA by downloading this free CAPA template.
A modern QMS can get you out of a reactive pose and into a proactive one. Greenlight Guru, which is equipped with CAPA management software, automates CAPA workflows to make it easier for companies to identify and address systemic quality issues.
![21 CFR Part 820 Labeling and Packaging Control]() 21 CFR Part 820 Subpart K—Labeling and Packaging Control
21 CFR Part 820 Subpart K—Labeling and Packaging Control
 21 CFR Part 820 Subpart K—Labeling and Packaging Control
21 CFR Part 820 Subpart K—Labeling and Packaging ControlWhat it covers: 21 CFR Part 820 Subpart K covers labeling and packaging control. This includes label integrity, inspection, storage, and operations.
How to comply: 21 CFR Part 820 Subpart K contains two subsections: one specific to device labeling and the other covering device packaging. Subsection §820.120 requires manufacturers to control these labeling activities:
-
Label integrity: Print and apply labels so that they remain legible and attached as the device is used.
-
Labeling inspection: Determine an individual who can examine the label’s accuracy before release.
-
Labeling storage: Store labels so that you can identify them and not mix them up.
-
Labeling operations: Control labeling and packaging processes, so you don’t mix labels up.
-
Control number: Ensure a control number, if applicable, follows the device through distribution.
Subsection §820.130 is a short subsection that focuses on device packaging. It requires the packaging and shipment of devices in a way that protects products from alteration or damage during “processing, storage, handling, and distribution.”
Common mistake: Applying blanket rules on device labeling. Avoid this mistake by looking to FDA precedent and see what specific labeling needs your particular device will require.
FDA writes at length about key considerations when labeling sterilized devices: label what is and what is not sterile in a package, provide directions for sterilization if it’s not pre-sterilized, offer instructions on how to open the package so that the contents remain sterile, and more.
The exact importance of labeling varies depending on the device. If you’re producing sterilized devices, for example, labeling is extremely important.
![21 CFR Part 820 Handling storage and distribution]() 21 CFR Part 820 Subpart L—Handling, Storage, Distribution, and Installation
21 CFR Part 820 Subpart L—Handling, Storage, Distribution, and Installation
 21 CFR Part 820 Subpart L—Handling, Storage, Distribution, and Installation
21 CFR Part 820 Subpart L—Handling, Storage, Distribution, and InstallationWhat it covers: 21 CFR Part 820 Subpart L covers handling, storage, distribution, and installation. This includes stipulations for compliant processes for storing and distributing products.
How to comply: 21 CFR Part 820 Subpart is split, as the subtitle implies, into four sections.
The first subsection, §820.140, focuses on handling and requires you to have procedures that prevent “mixups, damage, deterioration, contamination, or other adverse effects” during handling.
The second subsection, §820.150, focuses on storage:
-
Control storage areas and stock rooms so that you can prevent “mixups, damage, deterioration, contamination, or other adverse effects” and prevent the accidental use of “obsolete, rejected, or deteriorated product.”
-
Describe how you authorize the receiving and dispatching processes from storage areas and stock rooms.
The third subsection, §820.160, focuses on distribution:
-
Control how you distribute devices. Only devices you approve for release should actually be distributed. Review purchase orders and resolve issues before distribution. Check whether devices with expiration dates have expired.
-
Maintain records for your distribution.
The fourth subsection, §820.170, focuses on installation:
-
Create installation and inspection instructions for devices that require installation. Instructions have to guide users so that they use it as intended.
-
Ensure the person doing the installing does it correctly and documents the installation.
Common mistake: Many medical device companies don’t define all the handling procedures that should be defined. If your product requires installation at point of use, for instance, then you also need to define that. That includes defining installation requirements and specifications. All of this must be clearly documented.
These definitions should also come with acceptance criteria to help users verify the correct installation. You’ll identify this criteria when defining user needs during design and development.
![21 CFR Part 820 Records]() 21 CFR Part 820 Subpart M—Records
21 CFR Part 820 Subpart M—Records
 21 CFR Part 820 Subpart M—Records
21 CFR Part 820 Subpart M—RecordsWhat it covers: 21 CFR Part 820 Subpart M covers records. This includes a set of general requirements, regulations for the quality system record and complaint files, as well as the oft-confused device master record and device history record.
How to comply: 21 CFR Part 820 Subpart M is one of the more complex sections of 21 CFR Part 820 and contains five subsections.
Subsection §820.180 lays out general requirements:
-
General: Maintain your records in such a way that they’re accessible to you and to FDA employees doing inspections.
-
Confidentiality: Mark records you don’t want anyone but FDA reading as confidential.
-
Record retention period: Retain records for the expected life of the device, “but in no case less than 2 years from the date of release for commercial distribution.”
Subsection §820.181 explains the requirements for the device master record (DMR). Manufacturers must maintain a DMR that includes:
-
Device specifications
-
Product process specifications
-
Quality assurance procedures and specifications
-
Packaging and labeling specifications
-
Installation, maintenance, and servicing procedures and methods
Subsection §820.184 explains the requirements for the device history record (DHR). Manufacturers must maintain a DHR that includes:
-
Dates you manufactured the device
-
The quantity of devices you manufactured
-
The quantity of devices you released for distribution
-
Acceptance records that show you manufactured your device in accordance with your DMR
-
The primary identification label and labeling you used for each production unit
-
Any device identifications and control numbers you used
Subsection §820.186 explains the requirements for a quality system record (QSR). Your QSR must include everything required in this part of 21 CFR Part 820, including §820.20.
Subsection §820.198 focuses on the requirements for complaint files:
-
Receive, review, and evaluate complaints and maintain them in files.
-
Review complaints to determine whether you should investigate.
-
Review, evaluate, and investigate any complaint that involves your device or its labeling and packaging failing.
-
Determine a person or persons who will report complaints that FDA requires you to report under Part 803.
-
Document investigations performed due to the requirements in this subsection.
-
Make complaints accessible if the complaint unit is separate from your main manufacturing establishment, including if the unit is outside the United States.
Download our free complaint template that’s designed to help you streamline the complaint process.
Common mistake: : Some companies log everything as “product issues” when, in fact, many of them should be logged as complaints. One company logged over 500 product issues, but many of these issues mentioned problems, like melting, burning, and smoking, that went beyond issues. These are complaints—not issues.
This lack of proper logging increases the likelihood that a company will receive a warning letter. You can reduce the chance of these citations by using a quality management solution with purpose-built complaint management workflows that flag high risk situations.
![21 CFR Part 820 Servicing]() 21 CFR Part 820 Subpart N—Servicing
21 CFR Part 820 Subpart N—Servicing
 21 CFR Part 820 Subpart N—Servicing
21 CFR Part 820 Subpart N—ServicingWhat it covers: 21 CFR Part 820 Subpart N covers servicing.
How to comply: Not every device requires servicing but those that do need to comply with this section:
-
Maintain instructions for performing and verifying servicing if it’s a requirement for your device.
-
Analyze service reports.
-
Identify service reports that Part 803 requires you to report as complaints.
-
Document all service reports.
Common mistake: Some medical device companies lay out instructions for servicing but don’t do enough to verify the device meets specifications after said servicing. To ensure specifications are met, define your servicing requirements, specifications, and procedures. Maintain records to confirm that servicing was done correctly, and was in fact verified as effective.
Even if you outsource servicing to suppliers, these requirements hold true. Qualify, evaluate, and monitor the servicing suppliers you use and list them on your approved supplier list (ASL).
![21-cfr-part-820-subpart-o-statistical-techniques]() 21 CFR Part 820 Subpart O—Statistical Techniques
21 CFR Part 820 Subpart O—Statistical Techniques
 21 CFR Part 820 Subpart O—Statistical Techniques
21 CFR Part 820 Subpart O—Statistical TechniquesWhat it covers: 21 CFR Part 820 Subpart O covers statistical techniques. Compliant statistical techniques are intended to help manufacturers establish, control, and verify the “acceptability of process capability and product characteristics.”
How to comply: Statistical techniques are a core part of the service reports in 21 CFR Part 820 Subpart N and receive their own set of requirements for:
-
Identifying valid statistical techniques to implement for studying “process capability and product characteristics.”
-
Base sampling on valid statistical rationale.
Common mistake: FDA leaves the scope of statistical techniques up to the medical device establishment. Keep abreast of innovative statistical techniques to give your company a leg up for performing clinical trials and making benefit-risk decisions.

![Greenlight Guru is a 21 CFR Part 820 Compliant QMS]() Simplify 21 CFR Part 820 Compliance with a Medical Device QMS Solution
Simplify 21 CFR Part 820 Compliance with a Medical Device QMS Solution
 Simplify 21 CFR Part 820 Compliance with a Medical Device QMS Solution
Simplify 21 CFR Part 820 Compliance with a Medical Device QMS SolutionFollowing the FDA quality system regulations outlined in 21 CFR Part 820 can be a simple or challenging task depending on the type of QMS solution used by the manufacturer.
Legacy and ad hoc systems create burdensome hoops that teams must jump through to satisfy compliance requirements, contributing in many of the common mistakes mentioned throughout this guide.
Modern QMS solutions help manufacturers do their jobs better while eliminating work. No more fiddling with your QMS to ensure it aligns with regulations: Greenlight Guru’s QMS aligns with the latest industry regulations and standards, including 21 CFR Part 820 and 21 CFR Part 11, ISO 13485, and ISO 14971. It is the only industry-specific QMS software that allows medical device companies to achieve total lifecycle traceability within a closed-loop quality system.
With Greenlight Guru, you can finally move beyond baseline compliance and achieve True Quality outcomes for your quality system and the medical device it’s meant to support. Get your free demo of our QMS software today.
Free eBook: Ultimate Guide to 21 CFR Part 820 — FDA's QSR for Medical Devices

%20Ultimate%20Guide%20to%2021%20CFR%20Part%20820%20%E2%80%94%20FDA%20Quality%20System%20Regulation%20(QSR)%20for%20Medical%20Devices.png?width=180&name=(cover)%20Ultimate%20Guide%20to%2021%20CFR%20Part%20820%20%E2%80%94%20FDA%20Quality%20System%20Regulation%20(QSR)%20for%20Medical%20Devices.png)
%20Ultimate%20Guide%20to%2021%20CFR%20Part%20820%20%E2%80%94%20FDA%20Quality%20System%20Regulation%20(QSR)%20for%20Medical%20Devices.png?width=250&height=324&name=(cover)%20Ultimate%20Guide%20to%2021%20CFR%20Part%20820%20%E2%80%94%20FDA%20Quality%20System%20Regulation%20(QSR)%20for%20Medical%20Devices.png)







