
On Feb. 2, 2026, the Quality Management System Regulation (QMSR) officially replaced the Quality System Regulation (QSR). The QMSR incorporates ISO 13485:2016 by reference, harmonizing the standard with the US regulation.
One consequence of harmonization, however, is that the old Quality System Inspection Technique, known as QSIT, also was retired on Feb. 2nd. FDA has created a new document in it’s place: the updated Inspection of Medical Device Manufacturers Compliance Program (CP), numbered 7382.850.
The program describes how FDA investigators will evaluate device manufacturers under QMSR, and while inspections themselves will remain very similar to what manufacturers experienced under QSR, there are changes that medtech companies need to be aware of going into their next inspection.
Fortunately, we have both the Compliance Program itself and a recent FDA town hall to help us better understand the changes. In this article, we’ll take a look at the new Compliance Program, what’s changed, and how you can use it to prepare for your next inspection.
What changed when QSR became QMSR
At the FDA’s April 1, 2026 town hall on the program, Karen Masley-Joseph, Senior Advisor in FDA's Office of Medical Device and Radiological Health Inspectorate, put it plainly: the agency is no longer using QSIT, and the new Compliance Program now contains the inspection process for medical device manufacturers.
The QMSR brings ISO 13485:2016 into Part 820, along with Clause 3 of ISO 9000:2015, and adds FDA-specific provisions where the agency needed to preserve existing statutory requirements. The legal effect, as the Compliance Program states, is that the referenced standard is treated as though it were printed in the Code of Federal Regulations. Aside from where it differs from the CFR, ISO 13485 has carried the force of law since the rule became effective.
Two major themes shaped the inspection process. The first is an emphasis on total product life cycle, meaning the agency assesses a device from design and development through postmarket surveillance. The second is benefit-risk, meaning compliance and enforcement decisions account for the device's benefits and risks to patients, including patient perspectives where reliable information is available.
The inspection map: six QMS areas and four OAFRs
The clearest way to understand a 7382.850 inspection is the diagram at the center of the program.
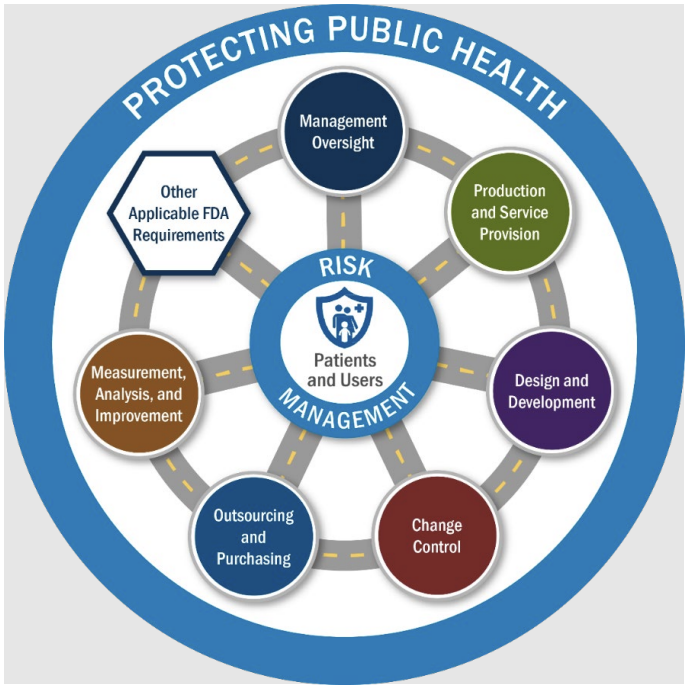 Source: CP 7382.850 Inspection of Medical Device Manufacturers Part III, Diagram 1
Source: CP 7382.850 Inspection of Medical Device Manufacturers Part III, Diagram 1
Patients and users sit in the middle. A ring of risk management surrounds them, reflecting FDA's emphasis on using a manufacturer's own risk management documentation as the focus of the inspection. Around that, the program organizes the QMSR requirements into six quality management system (QMS) areas and four Other Applicable FDA Requirements (OAFRs).
The six QMS areas are:
-
Change Control, covering changes to the QMS, software, products and processes, and purchasing
-
Design and Development, from design inputs and outputs through verification, validation, software validation, and transfer
-
Management Oversight, including the quality management system itself, management review, the medical device file, planning of product realization, and the risk-based approach required by Clause 4.1.2(b)
-
Measurement, Analysis, and Improvement, including complaint handling, feedback, internal audits, analysis of data, control of nonconforming product, corrective action, and preventive action
-
Outsourcing and Purchasing, covering outsourced processes and the purchasing process
-
Production and Service Provision, including process validation, identification and traceability, and, for sterile products, sterilization and sterile barrier systems
The four OAFRs are:
-
Medical Device Reporting, under 21 CFR Part 803
-
Reports of Corrections and Removals, under Part 806
-
Medical Device Tracking Requirements, under Part 821
-
Unique Device Identification, under Part 830
Each QMS area and OAFR breaks down into multiple elements, and each element ties to one or more specific requirements, mostly ISO 13485 clauses with QMSR additions layered in. Attachment A of the Compliance Program maps all of this in tables, and it’s perhaps the most useful part of the document for a quality team trying to anticipate what an investigator will examine.
Beyond the areas and OAFRs, investigators also review a set of general items on most inspections:
-
Registration and listing
-
Marketing authorizations
-
Any observations from previous Form FDA 483s or open compliance issues
-
Anything specified in the inspection assignment.
How an inspection unfolds
Much of the day-to-day experience of an inspection under the new Compliance Program will feel familiar to those who have gone through inspections under QSIT. As Masley-Joseph described at the town hall, a domestic inspection still opens with an FDA 482 Notice of Inspection. Investigators still tour the facility, ask how products and processes work, discuss roles and responsibilities, review quality data and documents, interview staff, and observe processes in action. If they find significant problems, they still issue an FDA 483 listing the observations, and the manufacturer can still annotate the form or respond in writing afterward.
Much of what’s changed is the increased emphasis on risk management. The investigator will first become familiar with the manufacturer's roles, products, and processes, then identifies the product risks that could adversely affect patients or users. Throughout the inspection, the investigator reviews the manufacturer's risk management documentation to understand those risks and how they are controlled. From there, the investigator selects an element within a QMS area or OAFR and evaluates the related requirements. The areas do not have to be examined in any set order, and if the inspection surfaces objectionable conditions, the investigator can add elements as needed.
.png)
Two inspection models
Every inspection follows one of two models, and the model determines how much of the quality system gets examined at a minimum.
Model 1: broad coverage across the whole system
Inspection Model 1 is the broad-coverage approach. The investigator identifies product risks, then evaluates at least one element from each of the six QMS areas, plus the applicable OAFRs and general items. Every main part of the quality system is touched on every Model 1 inspection, even if it’s only one element.
Model 1 applies to non-baseline surveillance, compliance follow-up, for-cause, specific product risk assignment, and PMA postmarket inspections.
Model 2: deeper coverage of named elements
Inspection Model 2 goes deeper. Instead of one element per area, it names specific minimum elements the investigator must evaluate across all six areas. Design and Development alone expands to inputs, outputs, review, verification, validation, software validation, and transfer. Measurement, Analysis, and Improvement expands to analysis of data, control of nonconforming product, complaint handling, feedback, internal audits, corrective action, and preventive action.
Model 2 applies to baseline surveillance inspections and PMA pre-approval inspections, where the agency has either no inspection history to rely on or a pending application to approve. In both models, the investigator can always evaluate additional elements if conditions warrant.
The most important changes for Quality teams
Several changes carry real weight for how a manufacturer should prepare, and they go beyond the new diagram.
A greater emphasis on risk management
The QMSR requires manufacturers to document one or more processes for risk management in product realization under Clause 7.1, and to apply a risk-based approach to controlling QMS processes under Clause 4.1.2(b). Investigators will review risk management documentation throughout the inspection and use it to decide where to look.
This emphasis on risk also applies to administrative processes. Masley-Joseph explained that Clause 4.1.2(b) applies to all QMS processes, including functions like document control and training, yet manufacturers do not need to build separate formal risk assessments for those functions. Instead, decisions about an administrative process should reference the existing risk management documentation for the related product or process. A decision about how often to retrain staff on a manufacturing procedure, for example, should be informed by the documented risk of that procedure rather than by a standalone training risk assessment.
Training effectiveness follows the same proportionality. Asked about that topic in a separate question, Tonya Wilbon, Assistant Director for Postmarket Industry Education and Consumer Education, Center for Devices and Radiological Health (CDRH), explained that under the requirements in ISO 13485, the rigor of an effectiveness check should match the risk of the work. Higher-risk processes may call for supervisor evaluations, skill assessments, or performance monitoring, while simpler verification, like quizzing, can be enough for lower-risk work.
Records that QSIT could not touch are now in scope
Under the old QSR, management review records, internal audit reports, and supplier audit reports were generally exempt from FDA review. That exemption is gone under the QMSR. Keisha Thomas of CDRH's Office of Product Evaluation and Quality traced the change to the rule's preamble and explained how it plays out: for baseline surveillance and PMA preapproval inspections, these processes are expected to be reviewed, and for other inspection types they may be reviewed depending on what the investigator selects.
She noted that records predating the effective date are still fair game for review if the inspection leads there. A design deficiency behind a significant recall, for instance, could prompt an investigator to examine how the manufacturer audited its design and development process.
Culture of quality is part of what gets assessed
The QMSR preamble frames top management's responsibility in cultural terms, and the program quotes it: "A culture of quality meets regulatory requirements through a set of behaviors, attitudes, activities, and processes." However, this does not require a special exhibit for inspectors. Manufacturers do not need to take separate steps to document quality culture for an inspection. The culture shows up in the decisions made and actions taken across the quality system, particularly in how risk information feeds risk-based decisions.
What happens to records created before Feb. 2, 2026
A common worry is whether years of existing records suddenly fail the new rule. They do not. Masley-Joseph confirmed that manufacturers do not need to revise or recreate records made before the effective date, do not need to add ISO 13485 references to older documents, and do not need to scrub QSR-era terms such as design history file or device master record from historical records.
However, the agency expects manufacturers to identify where their processes need to change to meet the QMSR and to develop a plan for those changes. If a gap analysis of pre-QMSR records reveals a real deficiency, such as feedback that was never fed into the risk management process, the manufacturer should develop and implement a plan to close it. Old records do not need new labels, but old gaps still need correction.
After the inspection: classifications and regulatory action
Part V of the Compliance Program covers what happens once the investigator leaves. Inspections receive one of three classifications:
-
No Action Indicated (NAI): no objectionable conditions were observed.
-
Voluntary Action Indicated (VAI): objectionable conditions were documented but do not meet the threshold for regulatory action.
-
Official Action Indicated (OAI): objectionable conditions were supported by evidence and regulatory action is recommended.
The Compliance Program guides that decision with two situations.
-
Situation 1 lists examples that point toward an initial OAI classification, such as a failure to establish or maintain a risk management process in product realization, or postmarket feedback that never feeds back into risk management.
-
Situation 2 lists examples that typically result in a VAI, where deficiencies indicate a low probability that nonconforming or defective product will reach patients. Both situations existed under prior versions of the program, and the examples were updated to reflect the QMSR.
Keep in mind, the threshold for taking action has not moved, and the regulatory requirements that determine when action is warranted are unchanged, even though the framework for evaluating compliance was updated to align with the QMSR and ISO 13485.
When action is warranted, the program lays out a ladder that runs from advisory measures such as untitled letters, warning letters, and regulatory meetings, through administrative actions including civil money penalties, administrative detention, citations, and recall authority under Section 518 of the Federal Food, Drug, and Cosmetic Act, to judicial actions such as seizure, injunction, and prosecution. Manufacturers generally have 15 business days after an inspection closes to submit their corrections and corrective action plans in writing, and prompt voluntary correction remains the outcome the agency prefers.
Preparing for a risk-based inspection
Wilbon's guidance at the town hall was that the best way to prepare is to meet the QMSR requirements that apply to the product, and to be ready to discuss and produce records that demonstrate that compliance.
Given the rule's emphasis on integrating risk management across the lifecycle, she suggested a specific exercise: make sure your internal audit program evaluates how risk management is actually integrated through product realization and whether a risk-based approach controls the QMS processes. That means tracing risk controls through the system, starting with an identified patient risk and following its controls through each process they touch, then being ready to show that risk-based decisions were justified, documented, and acted on.
A team that can trace a single patient risk from design inputs, through process validation and production controls, into complaint handling and postmarket feedback, and back into risk management, is demonstrating exactly the connected, risk-driven quality system that 7382.850 is built to evaluate.
See how your quality system holds up under the new Compliance Program
A risk-based investigation follows the connections between processes, from an identified patient risk, through design and production controls, into complaint handling and postmarket feedback, and back into risk management. Quality systems that hold those links together come through inspection in good shape. The ones that keep risk management in a separate file are where the new program finds its observations.
Greenlight Guru is the QMS purpose-built for medical device manufacturers, with design controls, risk management, CAPA, supplier management, and postmarket data connected in one validated system aligned to ISO 13485 and the QMSR.
If your team is preparing for its first inspection under the new program, get your free demo of Greenlight Guru to see how you can keep risk management connected throughout your QMS.
Your QMSR Readiness Playbook

.png?width=575&name=Quality%20Lead%20Magnet%20Slide-in%20(3).png)
.png?width=250&height=321&name=Quality%20Lead%20Magnet%20Slide-in%20(3).png)



