
Nobody starts a medtech company to manage documents, but oddly enough, that's exactly where a lot of pre-market teams spend huge amounts of time.
The problem usually grows quietly. Your engineers are focused on product development, and if you have a QA lead, they may find spreadsheets to be manageable early on. But the issues with this setup compound quickly. As the team grows, design iterations start to accumulate, and the pressure to hit timelines increases, your documentation naturally outgrows your ability to maintain it. By the time you realize there's an issue with your QMS, it's become systemic, and it's significantly more expensive to fix than it would have been six to twelve months earlier.
This guide is written for pre-market medtech teams that want to move fast and are questioning whether their current setup will hold at scale.
Compliance isn't the bottleneck. Manual compliance is. There is a meaningful difference between the two, and it's what this guide is about.
What follows is a practical look at where quality debt accumulates, what it costs in real terms, and what a right-sized QMS actually looks like for a lean pre-market team.
BONUS RESOURCE: Click here to download your free content toolkit for product development!
Table of contents
1. The timeline reality check
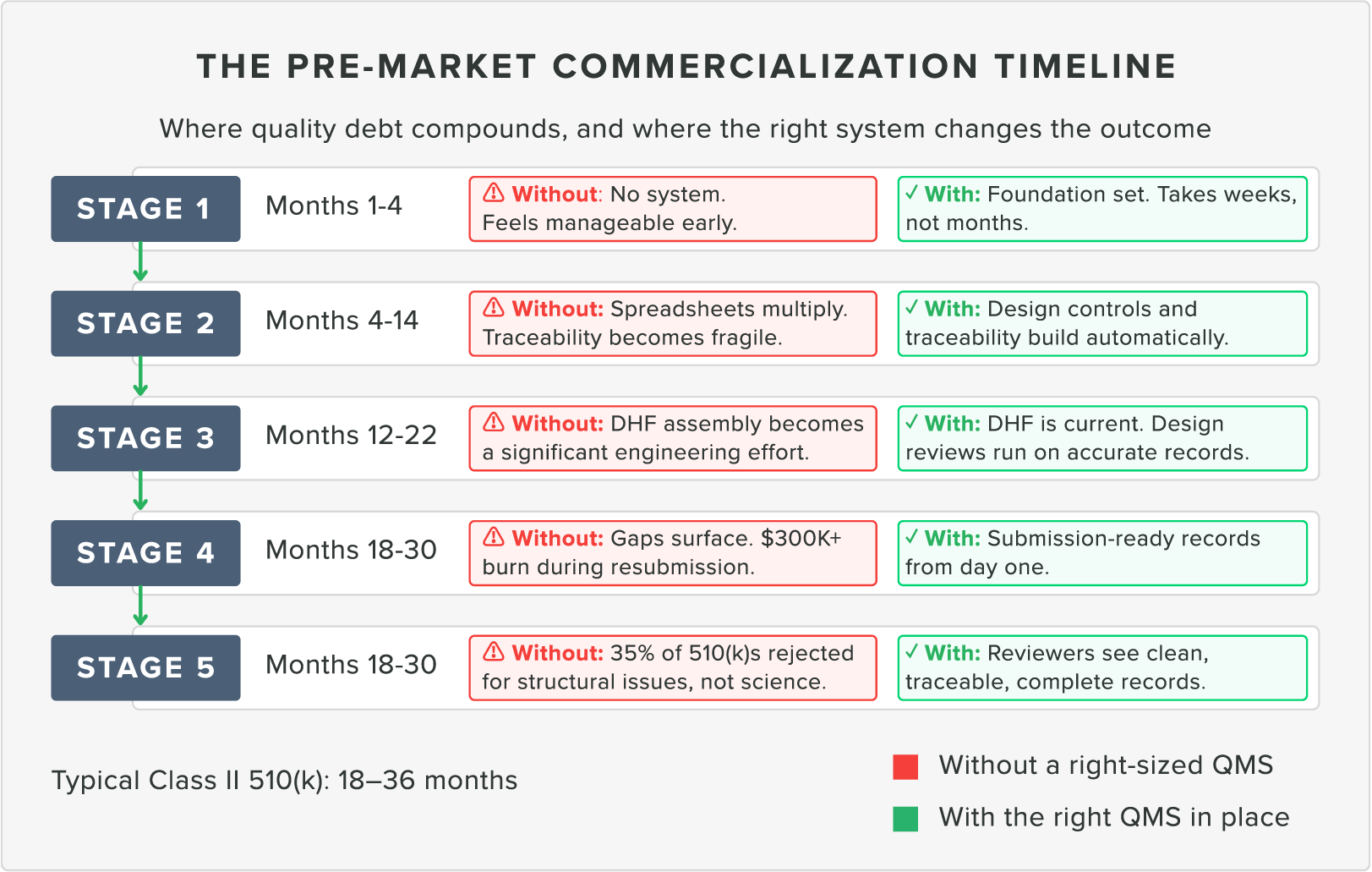
A 510(k) typically takes 18 to 36 months from working prototype to cleared device.
The place where quality debt becomes expensive is not at submission. It's in the 6 to 12 months before submission, when teams discover that their system isn't fully traceable and ready for submission, let alone an audit.
Why "we'll handle it closer to submission" is worth examining
It's a reasonable instinct. In the beginning, submission feels like it's miles away. The problem is that getting your QMS in order is a task that only gets larger as you ignore it. A team that implements an eQMS early might spend weeks on setup. A team that implements late could spend months on cleanup.

Cleanup always costs more than setup. The only variable is when you pay, and at what rate.
2. What your current system is actually costing you
The direct cost of spreadsheets and shared drives is close to zero, which is why most medtech companies start off using them. The indirect cost shows up in engineering hours spent on administration, QA time consumed by approval chasing, and delays in commercialization.

Run the numbers on your own team
Here are some questions worth answering:
How many hours per week are your engineers spending on documentation tasks that aren't actual engineering work?
How much QA time goes toward chasing approvals, rebuilding traceability, or reformatting records for design reviews?
How confident are you that your DHF, today, could be presented to an FDA inspector within two weeks?
When did your last design review take longer than expected because the underlying records were incomplete or misaligned?
But what's the ROI?
To get a better sense of the ROI on an eQMS, try out our ROI calculator.
Try our free, fully customizable ROI calculator today!
Two patterns that show up consistently

The question worth asking isn't whether your team can afford a right-sized QMS. It's whether you can afford six more months of delay and the engineering time required to clean up what the current approach is accumulating.
3. The Quality and Engineering gap
In most pre-market medtech companies, Quality and Engineering are managing the same product from different systems, different workflows, and often different versions of the same documentation.
From disconnected to streamlined

What this tension sounds like in practice

Neither team is wrong. The friction is a predictable outcome of systems that weren't designed to work together.
4. What Right-Sizing Actually Means at Each Stage
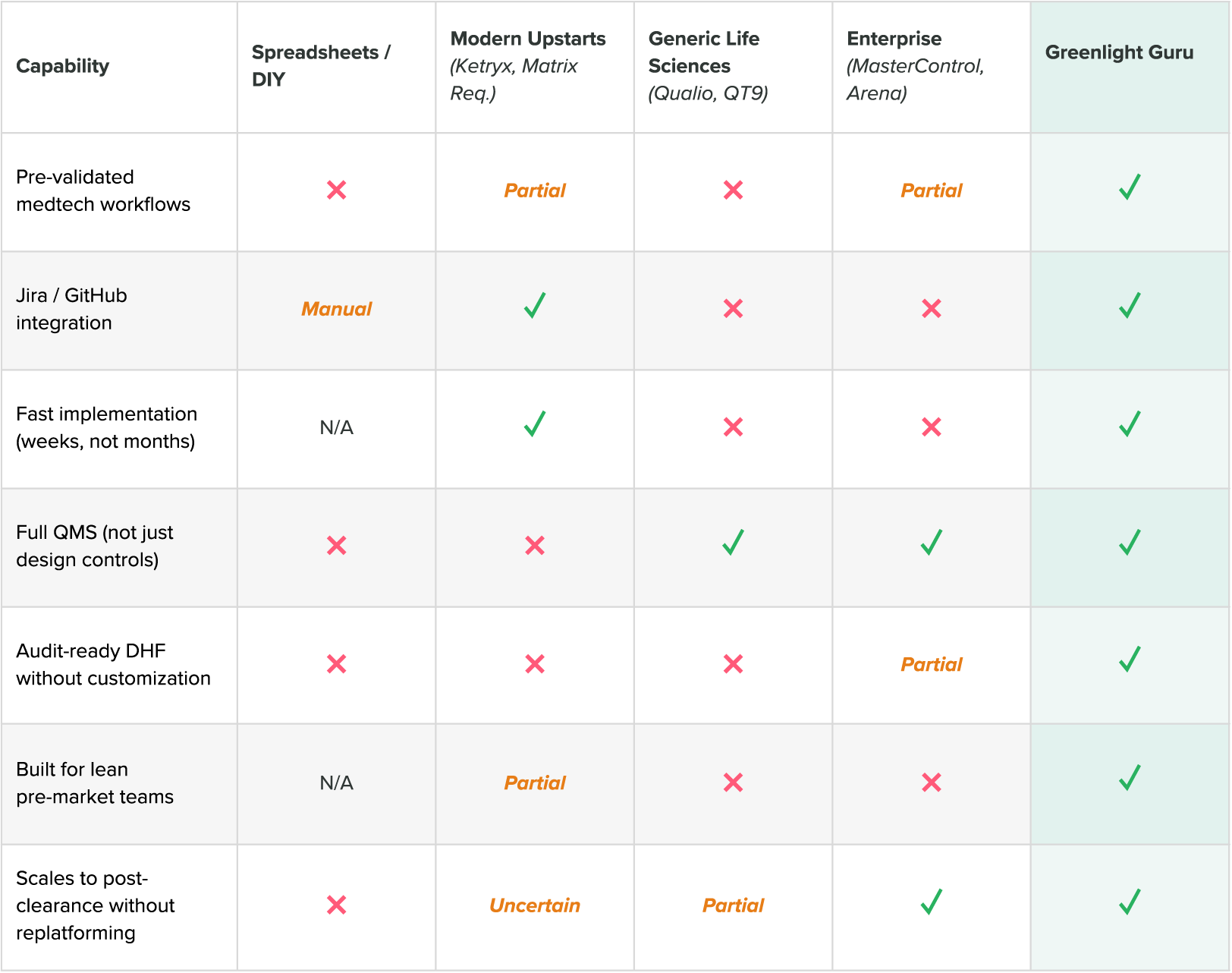
What right-sized looks like at each development stage
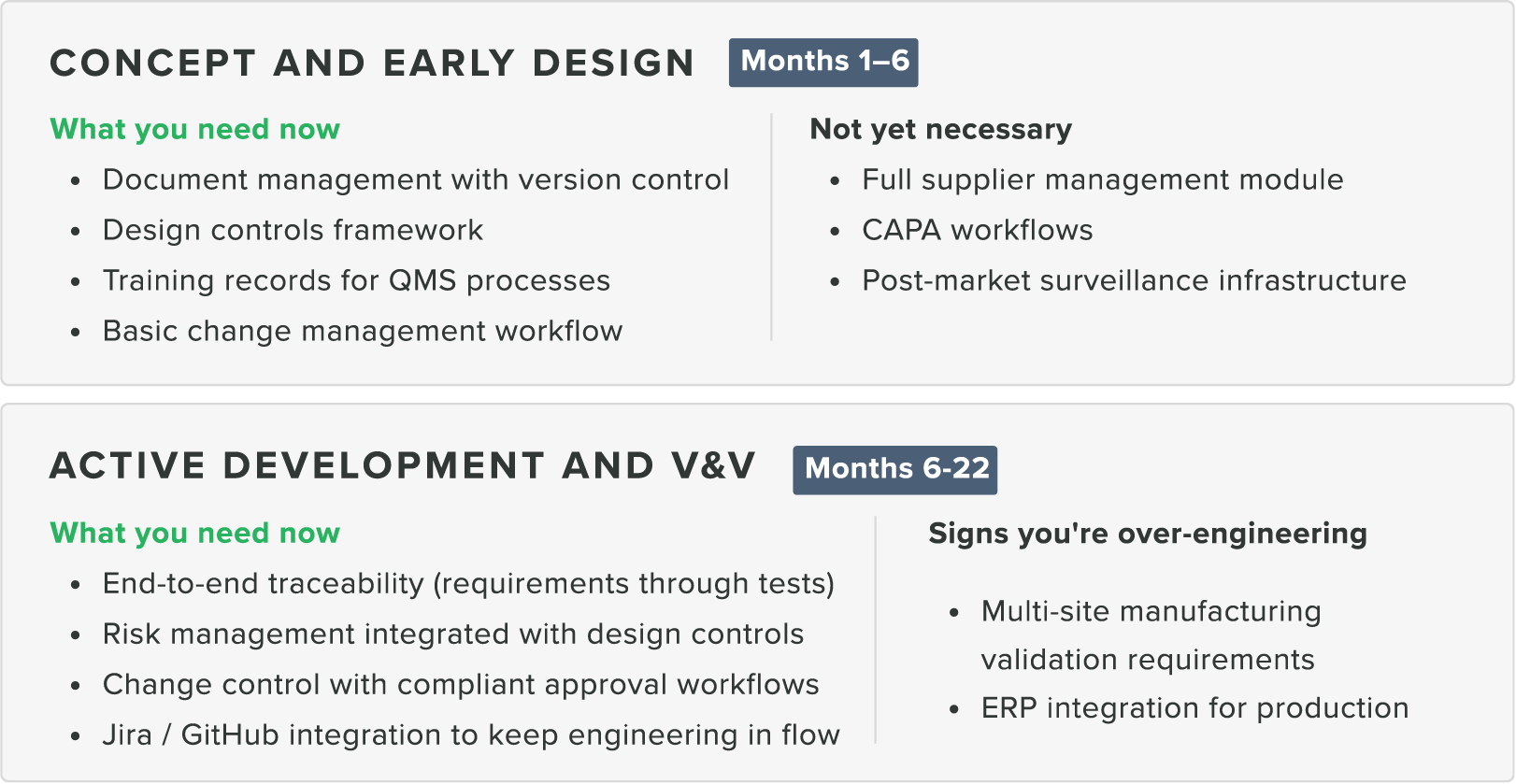
Can your system do this?
Design inputs are formally documented and version-controlled
Every requirement is linked to at least one verification test (bidirectional traceability)
Change control records show who approved what, and when
Your DHF could be assembled and presented to an inspector within two weeks
Risk records update automatically when requirements change
Engineers can complete compliance tasks without leaving their primary development tools
QA has real-time visibility into DHF completeness without manual reporting
Document approval workflows do not rely on email
Your QMS is validated and audit-ready
A new team member can be onboarded to the system in under a week
If three or more of those are unchecked, your current approach is creating cleanup you'll pay for before submission.
5. What good looks like and questions to ask

Questions worth asking in every evaluation
How much ongoing engineering time does your system require after implementation, week over week?
Can quality and product teams work in the same environment without a manual handoff step between them?
How long does a standard implementation take, from kickoff to audit-ready?
Walk me through what happens to our traceability records when we make a late-stage design change.
How does DHF assembly work? Show me in the live product.
Which FDA QMSR and ISO 13485 workflows are pre-validated versus built from scratch?
Can you show me how a Jira ticket connects to a design requirement in the live product, right now?
6. How to make the case internally




7. Getting started without losing momentum
The number one concern we hear from pre-market teams considering the switch to an eQMS is that the implementation itself will be the bottleneck.

Teams that implement a right-sized QMS early don't slow down. They stop accumulating the documentation debt that will create a significant slowdown before submission.
What customers say about the transition

You don't need a full QA team to start
One of the more persistent myths about pre-market QMS implementation is that you need a dedicated quality hire before you can stand up a real system.
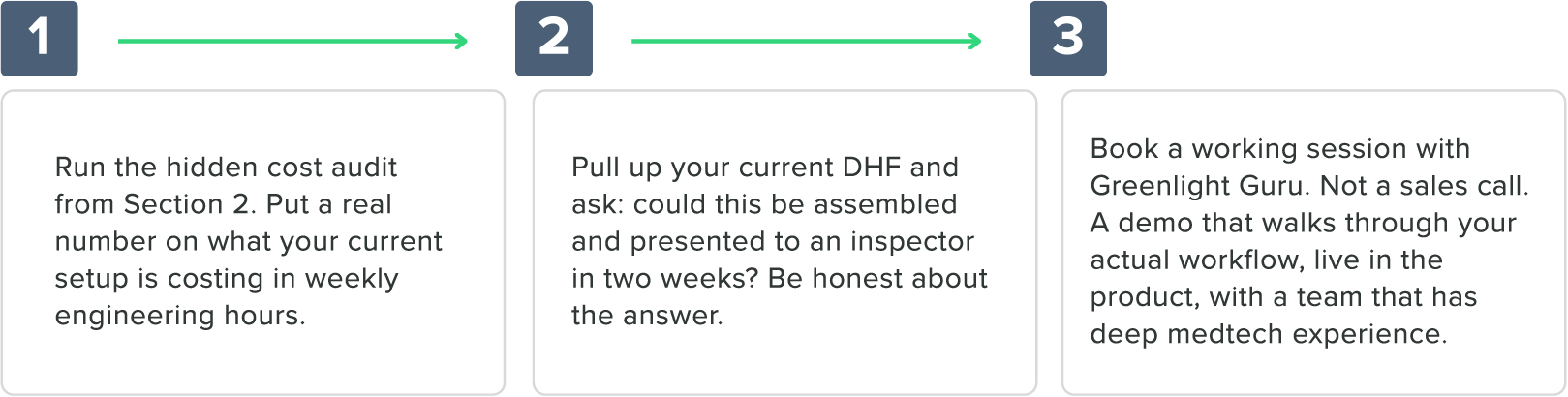
Three concrete next steps
8. Frequently asked questions
When should a medical device startup implement a QMS?
Earlier than most teams do. The ideal window is as soon as you have a defined device concept and a small team actively working on design and development.
How long does it take to prepare a 510(k) submission?
The submission package itself typically takes 2 to 6 months to compile, but that assumes your documentation is organized, your traceability is complete, and your DHF is current.
What is a right-sized QMS for a pre-market team?
A right-sized QMS provides enough structure to meet ISO 13485 and FDA QMSR requirements without adding administrative overhead to an engineering team.
Can we manage design controls in spreadsheets?
Spreadsheets work as a short-term documentation tool when your design is relatively simple. They become problematic when you have more than a few design inputs, multiple design iterations running concurrently, more than two engineers working in the system, or a design review that requires current and accurate traceability.
What is the fastest way to build a compliant DHF?
Build it as you go, not after the fact. A DHF assembled in real time is faster to produce and more defensible at review than one reconstructed from memory and archived files.
How does QMSR affect pre-market teams?
The FDA's Quality Management System Regulation, which aligned with ISO 13485 in 2026, raises the structural expectations for quality systems at every stage of device development.
Do we need a full-time QA hire before implementing a QMS?
No. Most pre-market teams implement their first QMS with a part-time QA resource or a regulatory consultant.
BONUS RESOURCE: Click here to download your free content toolkit for product development!
Ready to see what it looks like in practice?
A right-sized QMS is great in theory, but without the right QMS solution, your team may still end up drowning in manual documentation while missing or outdated records leave you vulnerable to audit findings.
Greenlight Guru is purpose-built to be a lightweight, flexible eQMS that keeps early-stage teams compliant from the start. Our eQMS combines easy implementation with powerful medtech-specific modules like design controls, risk management, supplier management and more. And because it integrates with the tools engineers love, like GitHub and Jira, you won't have to worry about work happening outside the system that isn't documented.
If you're ready to stop compromising on your QMS solution, then get your free demo of Greenlight Guru today!
Content Toolkit for Product Development






