
The US Food and Drug Administration (FDA) has released the quality system inspection data for citations of medical device manufacturers in 2020.
Although FDA cited manufacturers for a wide range of issues, three areas in particular stand out. Together, citations for CAPA, complaints, and design controls made up a whopping 35% of all FDA 483 citations issued in FY2020.
And this is by no means the first year these three areas have topped the citation charts. CAPA, complaints, and design controls have consistently been the top three cited areas for over a decade.
Given these numbers, it’s clear that companies are having trouble following the quality system regulations surrounding these three subsystems. So, we decided to take a closer look and help medical device manufacturers stay proactive and avoid any related citations during your next FDA inspection.
CAPA Citations — defining and following risk-based procedures
Corrective and preventive action (CAPA) violations were cited 197 times, making CAPA the most cited area by FDA in 2020.
Of those, the vast majority—165 citations—were issued to manufacturers who did not document their CAPA processes. Thirty-two citations were issued to manufacturers who failed to adequately capture their CAPA activities.
The number of CAPA citations is concerning because CAPAs are meant to be initiated in response to systemic issues with a medical device. These are issues that have the potential to become major problems and negatively affect the health and safety of patients. It’s unsettling that so many citations were issued as a result of manufacturers’ failure to have a documented process for CAPA.
So, the biggest takeaway from those numbers is that you absolutely must have a documented process for CAPA, and you must also document your use of that process. To FDA, if it wasn’t documented, it didn’t happen.
And if you’re not sure about the efficacy of your CAPA process, here are some high-level steps I’d recommend you have in your process:
-
Identify existing and potential causes of nonconformities and/or quality issues.
-
Investigate the root causes of any nonconformities relating to product, process, or the quality system.
-
Review the need for, and identify, action(s) needed to correct and prevent recurrence of nonconforming product or other quality issues.
-
Verify or validate that the corrective action does not adversely affect the finished device.
-
Specify a corrective and/or preventive action plan, including roles and responsibilities, and a plan for a verification of effectiveness.
-
Document, review, and approve the CAPA activities.
-
Verify and document the effectiveness of the CAPA activities.
Interestingly, CAPAs are often initiated due to complaints that manufacturers receive once their devices are on the market—which means it’s likely not a coincidence that the second-most cited area in 2020 was complaints.
Complaint Citations — handling and investigating complaints
Claiming the dubious honor of second place—with 193 citations from FDA—is complaints.
For context, every one of the citations issued by FDA was against the process that manufacturers had (or failed to have) in place for handling complaints. FDA issued 139 citations for failure to document processes or maintain files.
The remaining citations were issued for failure to investigate complaints, a lack of rationale for not investigating complaints, and a failure to promptly investigate complaints that were MDR reportable.
I hope you’re beginning to see a trend here. Just like with CAPA citations, there was a failure among some manufacturers to document their complaint processes. Perhaps even more startling, though much less common, were the citations for failing to properly investigate complaints.
Regardless of the specifics of all these citations, the reality is, you absolutely need to have a defined and documented process for handling and investigating complaints. As you can see from the flowchart below, a good complaint handling procedure has a number of steps, all of which must be documented:

Hopefully, this chart also makes clear the connection between handling complaints and initiating CAPAs. A good complaint handling process will also include your CAPA procedure.
Design Control Citations — validating design and revalidating changes
Design controls are a little different from the first two areas because there are 10 subsections within 21 CFR Part 820.30 that FDA can issue citations against. However, out of the 178 times design controls were cited in 2020, 48 of them concerned design validation and 26 concerned design change. These two areas, which are closely related, make up just over 40% of all design controls citations.
Design validation requires that manufacturers ensure the device they have created meets its intended use or user needs. When a device undergoes a change—for instance, as a result of a CAPA investigation—that change must be revalidated.
There were a range of issues with validation, such as lack of procedures as well as a lack of documentation of activities such as software validation. In some cases, validation studies did not use production equivalent devices. For design change, the 26 citations were issued because manufacturers did not properly establish their procedures.
It’s worth noting that design controls are closely related to CAPA and complaints, too. You can think of their correlation to one another as a negative feedback loop:
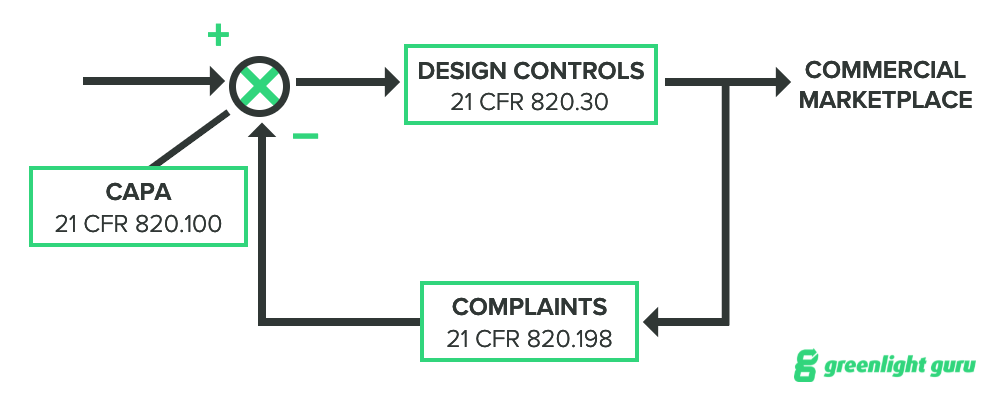
-
Complaints may trigger a CAPA.
-
If a CAPA is triggered, the outcome may affect design controls.
-
The manufacturer must validate any changes in the design.
-
The device returns to market.
Now, I should note that this example shows an entirely reactive feedback loop, and it is much better to take a proactive approach to both complaints and CAPA, but this diagram should illustrate how closely related these three areas really are within a quality system.
Improve your QMS processes with the only medical device success platform
By this point, I hope you’ve noticed that establishing proper processes and documenting their use is critical to leaving an FDA inspection without citation.
Often, the problem for medical device manufacturers isn’t that they don’t want to document their activities or processes, it’s that they’re using a hodge-podge of legacy tools to do so. Without a modern, purpose-built quality management system in place, it can be difficult to automate document management so that all records are kept up-to-date and compliance is maintained.
That’s why Greenlight Guru’s QMS software was designed specifically for the medical device industry. Our QMS has the latest FDA and ISO best practices built into every feature and offers Intelligent Document Management that provides seamless, closed-loop visibility and traceability.
Steer clear of citations at your next FDA inspection and stay proactive when it comes to the system you use to manage quality—get your free demo of Greenlight Guru today.
Looking for a design control solution to help you bring safer medical devices to market faster with less risk? Click here to take a quick tour of Greenlight Guru's Medical Device QMS software
FREE RESOURCE:
3-in-1 Quality Event Template Package
3-in-1 Quality Event Template Package






