











According to the original data findings of our 2020 State of Medical Device Product Development and Quality Management Report, over half of medical device manufacturers cited document control as a challenge.
Document control is the backbone of an effective quality management system (QMS). Both ISO 13485 and 21 CFR Part 820 require documentation and, if you are using an electronic system and approach, 21 CFR Part 11 outlines the administration of those documents.
Yes, you’ve likely heard the old adage: “If it isn’t documented, then it didn’t happen.” (And yes, I heard your eyes roll after reading that.) Yet, it’s true. Documentation is vital to every aspect and functional area of a medical device company. Documentation serves as a means to collaborate and communicate within your company. Documentation is critical to demonstrating compliance to regulatory bodies and auditing organizations.
Yes, regulatory agencies and external auditors take documentation seriously. In 2019, FDA issued 15% more inspection citations than the previous year.
Let’s put this in simple terms. The strength of your medical device company is directly related to how you manage documentation. Documentation is essential to your QMS. Documentation is an ISO and FDA requirement. Document control issues are still a very big challenge and all too common during FDA inspections and ISO audits.
The question is, if documentation is so important, then why do so many companies struggle with it?
This in-depth guide will take a deep dive into the problems that tend to afflict document control activities, provide detailed explanations of the procedures that make your documentation more efficient, and offer proven effective methods for implementing and leveraging a robust document management system that will catapult the success of your organization and medical device.
![#toc]() Table of Contents
Table of Contents
 Table of Contents
Table of ContentsDocument control structure
Document control terminology
FDA 21 CFR PART 820.40
ISO 13485:2016
EU MDR
21 CFR Part 11 Compliance is Essential
21 CFR Part 11 overview
Part 11 Validation
Relationship between Document Management and Document Control
Your document management system is working against your team
Why Document Management Gets a Bad Rap
Distinguish between document control myth and reality
5 Benefits of Document Management for Medical Device Companies
How to Regain (Document) Control within your QMS
Right-size your document control system as your company grows
Build a documentation strategy to guide your growth
Rebuild document management in phases when your company is established
Types of Document Control Systems
How Document Control Impacts Change Management
How Document Control Impacts Inspections and Audits
Optimize Document Control with a Medical Device QMS Solution

![#1]() What is document control?
What is document control?
 What is document control?
What is document control?Document control is a practice that organizations use to keep their teams accountable for tracking documents. Companies do this to ensure they follow high-quality processes and satisfy regulatory requirements.
In other words, document control refers to the policies and procedures medical device companies use to manage the many documents that flow through different departments, stakeholders, and third parties throughout the medical device product lifecycle.
You should want to document your work because ultimately, the quality of human lives is at stake. The documentation you have in place should support the safety and efficacy of your products. The documentation should demonstrate consistency, reproducibility, and repeatability of processes. The documentation should be an effective way of communicating what to do, when to do it, and how it was done.
Document control structure
Medical device companies run a multitude of different procedures to handle various inputs and outputs. Document control encompasses all of the policies that ensure that every single one of those procedures is accounted for and recorded. This includes how and when a procedure was executed and whether it was done in accordance with a company’s documented policy.
It’s helpful to think about document control in terms of a top-down hierarchy.
The top is your quality manual, which sets the company’s quality philosophies and policies; the next level down includes documented quality procedures and your device master record (DMR); the next level includes all documentation of forms, specifications, drawings, and work instructions; and the foundation of your documentation is made up of your records and files, which include quality records, design history files (DHF), and device history records (DHR).
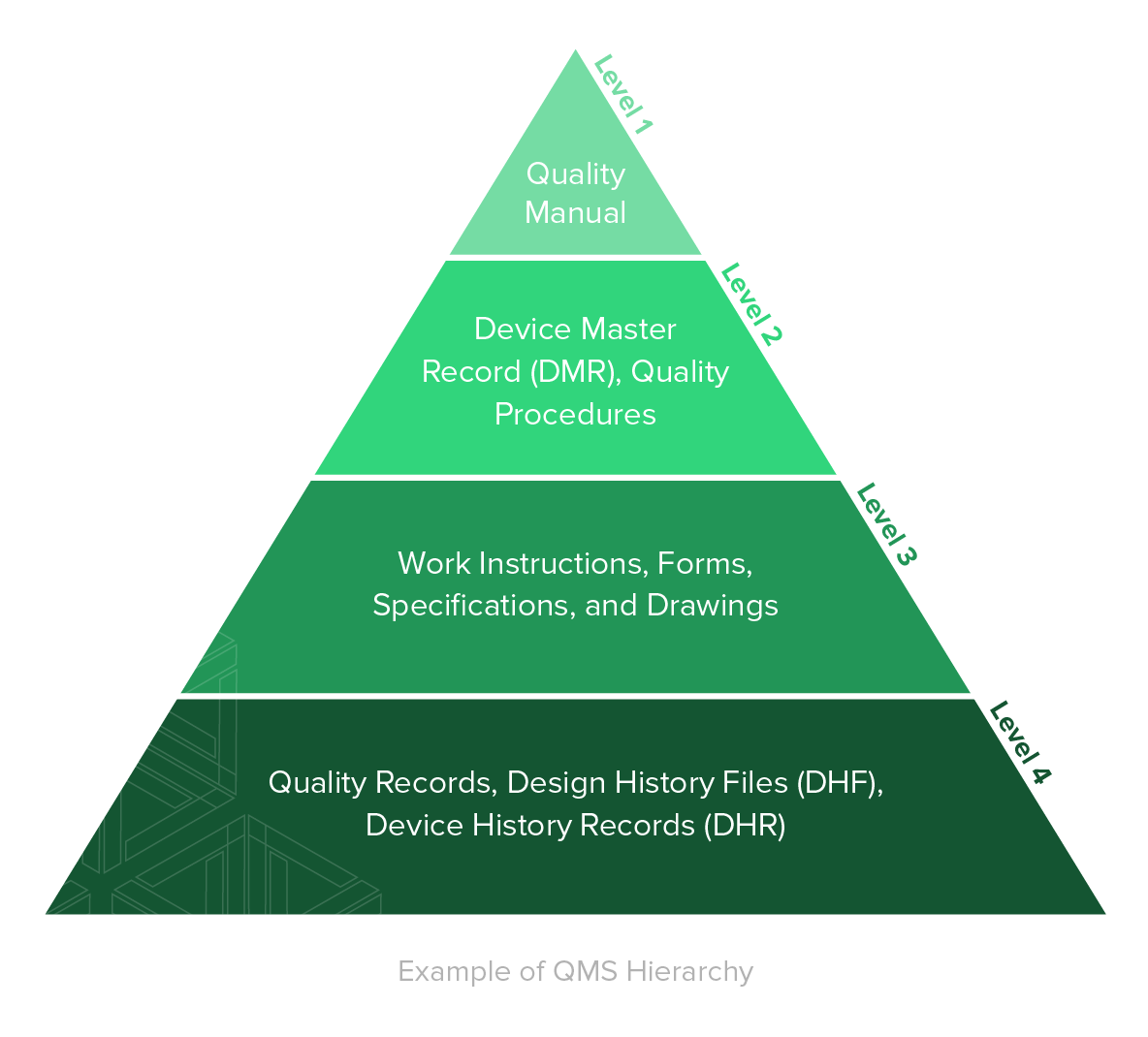 The hierarchy ranges from top level policies and procedures at the top of the pyramid to specific records at the bottom. The top level policies and procedures provide direction on how to do things within the QMS. The records at the bottom prove the employees are following those policies and procedures.
The hierarchy ranges from top level policies and procedures at the top of the pyramid to specific records at the bottom. The top level policies and procedures provide direction on how to do things within the QMS. The records at the bottom prove the employees are following those policies and procedures.
Document control terminology
In addition to what we’ve already covered in this guide, document control contains certain a number of key industry terms that are important to know and distinguish, which we list and define below.
-
Document control vs. document management: Some companies use the term “document management” instead of “document control”. Yes, these terms are very closely related. Think of “document management” as the system and process of managing and maintaining documentation. Think of “document control” as the means by which you generate, review, approve, and maintain revision history of the documentation.
-
Quality manual: A quality manual describes your company’s QMS, including the scope of it, a summary, exclusions from ISO 13485 (if any), the responsibilities and roles of your various team members, and more. The specifics of each quality manual is dependent on the size of the company.
-
SOP (Standard operating procedure): An SOP, generally, defines a specific process or procedure. In this context, a document control SOP focuses on the roles, responsibilities, procedures, and checks involved in compliant document control.
-
Change Order: A change order is a compilation of every decision you’ve made throughout the entire change management process pertaining to a certain document(s). Every department impacted by a particular change order will need to approve this.
-
Design history file (DHF): A DHF is a compilation of design and development records that show the design controls process was properly followed and documented throughout product development. You need to maintain a separate design history file for each type of device under development.
-
Device master record (DMR): A DMR contains or references all the information necessary to build and test your device. Each device master record has to adhere to FDA 21 CFR Part 820.181 and ISO 13485:2016 section 4.2.3 (referred to as “medical device file”).
-
Device history record (DHR): A DHR contains or references all the records that demonstrate that you manufactured a device according to your DMR. Your device history record needs to be maintained for every batch, lot, or unit.
-
Risk management file (RMF): An RMF contains or references all of your risk management activities, documentation, and records. You can organize it by an individual product or groups of products.
Understanding this medical device terminology is an important part of establishing your document control processes and integrating it into your QMS.
![#2]() Document control requirements
Document control requirements
 Document control requirements
Document control requirementsAs mentioned earlier, documentation is the backbone of an effective QMS and directly correlates to the strength of your medical device company.
Companies that can leverage good document control and easily prove compliance can afford to focus on product development, growth, and improving efficiencies. However, companies with poor document control always seem to be preparing for the next audit.
Let’s go over some of the most important requirements your document control system will have to be able to handle.
FDA 21 CFR PART 820.40
FDA's 21 CFR Part 820.40 pertains to document controls and requires medical devices to establish and maintain procedures to control all pertinent and applicable documents described throughout the FDA Quality System Regulations (QSR).
While the specific details of document controls defined by FDA is relatively short, the other parts of the QSR describe additional requirements for specific documentation necessary to effectively address the regulations.
ISO 13485:2016
ISO 13485:2016 is the globally harmonized standard for medical device quality management systems. There is significant overlap with FDA’s quality system regulation (QSR) and, generally speaking, compliance with one will set you up for compliance with the other.
The guidelines from ISO 13485 call for rigorous record-keeping. Section 4.2.4 (Control of documentation) in particular calls for document control that extends document maintenance through the life cycle of the device. And like the FDA QSR, additional documentation details are described throughout ISO 13485.
Important aspects of document control per FDA and ISO 13485 include:
-
Identifying the appropriate individual(s) to review procedures for adequacy.
-
Approvals shall include date and signature of the approvers.
-
Documents shall be available at all applicable points of use.
-
Obsolete versions to be removed for points of use.
-
Any changes shall be reviewed and approved by the same functions involved with the original review and approval.
-
Approved changes to documents shall be communicated to applicable users.
EU MDR
Europe’s new medical device regulation (EU MDR), which will go into effect on May 26, 2021 following a one-year delay of its initial enforcement date, comes with a host of changes relevant to document control.
EU MDR emphasizes a focus on the total product lifecycle, including post-market surveillance. The associated requirements necessitate a document control system that can produce items like a post-market surveillance report, which summarizes the results of all your post-market surveillance data, including any corrective actions.
Higher-risk devices may demand an even more rigorous documentation system to produce items, such as a periodic safety update report (as noted in Article 86), that require annual updates.
Article 25 of the regulation also presents major changes. MDR designates the “economic operator” status to distributors, importers, and EU-authorized representatives—each with its own documentation standards.
For more information on compliance with EU MDR, check out The Essential Guide To Preparing Your QMS For EU MDR.
![#3]() 21 CFR Part 11 compliance is essential
21 CFR Part 11 compliance is essential
 21 CFR Part 11 compliance is essential
21 CFR Part 11 compliance is essentialDoes FDA 21 CFR Part 11 apply to you? Chances are, probably so, unless your company is completely and entirely paper-based. In fact, a lot of companies try to avoid electronic records and signatures for fear of Part 11.
Realize this: If you send documents via email, have a server where electronic versions are stored, and/or sign with “wet” signature then scan, then you have electronic records and Part 11 is applicable.
When it comes to electronic document control, compliance with FDA’s 21 CFR Part 11 is essential. Part 11 regulates electronic documentation and electronic signatures. Being noncompliant is not an option if you intend to legally market your device in the United States.
21 CFR Part 11 overview
Part 11 of the Code of Federal Regulations Title 21 states that companies using electronic records shall “secure, computer-generated, time-stamped audit trails to independently record the data and time of operator entries, and actions that create, modify, or delete electronic records.”
In 1997, FDA issued and codified standards for the records and signatures they’ll accept. This applies to all FDA-regulated industries, such as pharmaceuticals and biotech, but our focus will remain on the applicability to medical devices.
Check out our FDA 21 CFR Part 11 guide to learn more.
eQMS skeptics argue that sticking to paper systems will be easier because using an eQMS means your systems must be subject to validation. In reality, validation isn’t as hard as skeptics make it out to be, especially when software from companies like Greenlight Guru is validated out of the box.
Part 11 Validation
Part 11 is part of 21 CFR Part 11, a set of FDA regulations that define acceptable criteria for electronic records and signatures.
Part 11 is notoriously confusing among medical device professionals. In 2003, FDA released a guidance document that clarified previous regulations and laid out requirements for software validation, audit trails, legacy system management, and record control.
FDA separates Part 11 into three sections: General Provisions, Electronic Records, and Electronic Signatures.
-
General provisions: This section covers the scope of Part 11, defines key terms, and outlines how companies can implement it.
-
Electronic records: This section establishes the requirements companies must follow to administer compliant electronic record-keeping systems.
-
Electronic signatures: This section covers requirements for electronic signatures, electronic signature components, and controls for codes and passwords.
Some of the confusion and stress from Part 11 comes from using the wrong tools.
A QMS that isn’t designed for the medical device industry will require re-engineering from the get-go. Since medical device manufacturing best practices and templates don’t come equipped in most general purpose systems, there will be added risks to account for every step of the way.
The more you have to customize your environment, the less dependable it becomes as requirements change. A custom environment means you have to configure and conduct every test yourself.
As your company evolves and regulatory standards change, you’ll need to regularly reconfigure and validate your QMS. This makes Part 11 validation intimidating.

relationship between document management and document control
Without a good system to manage it, your documentation becomes chaotic. The more chaotic your document control system becomes, the more likely the documents you complete will end up in a pile of other documents, leaving you with little control over the ability to trace, retrieve, and check them.
This isn’t the fault of your team, but rather the fault of legacy systems that work against your team. These legacy systems survive because of legacy myths—ideas that make them seem as effective as modernized solutions.
And this is where the concept of “document management” becomes very important. Think of document management as the system to manage all aspects of your company’s documents and records.
Document management is the core foundational element of every medical device company. A company’s success is entirely dependent on the quality and robustness of their document management system and practices.
Yet, sadly, all too many medical device companies fail to emphasize the importance of document management. Or maybe they choose to de-prioritize efforts on document management in lieu of other areas deemed more important.
Your document management system is working against you
You might have the best quality professionals available on your team, but without a good system, you can’t set them up for success. Your documents become information dumping grounds when quality professionals have nothing but ad hoc procedures and cumbersome practices.
Without a good system, quality professionals are stuck playing traffic cop, chasing down documents, and routinely interrupting team members to check and double-check whether a particular document was signed.
Plain and simple, it makes for bad team morale and worse document control.
Good document control allows your quality team to focus on strategic initiatives. Bad document control leaves quality professionals performing low value-add activities. And your document management system should help streamline and promote good document control.
Document control affects and is affected by people from engineering, marketing, operations and beyond. Each department needs to be assigned a role, a way in which they should utilize and leverage the company’s document control system. When that process is scattered, every additional hand makes it incrementally harder to manage.
Bad document management systems just don’t scale. Systems that don’t scale translate into an increase in resource allocation to manage an increase of documents. This creates a fragile, confusing, error-prone system that’s costly to staff and manage.
The topic of document management tends to be very polarizing to nearly all medical device professionals. Usually, there are two camps regarding document management. Those who embrace its importance. Or those who resist documentation and only yield because they have to in order to comply.
For the latter, I believe there are some underlying reasons, conventional wisdom, and/or historical practices that reinforce those feelings of animosity towards document management.
Why Document Management Gets a Bad Rap
Let me elaborate a bit on some of those negative underlying reasons some feel animosity towards document management.
First point of contention often relates to documentation for the sake of compliance. Don’t mishear what I’m about to say--yes, compliance is very important. However, if your reason for documentation is only for the sake of demonstrating compliance, then this misses the mark and intention.
I’ve seen countless cases where medical device professionals follow blindly and generate documentation to satisfy something a procedure states simply to check a box and satisfy an internal company requirement. I personally have worked within these types of environments before.
Filling out a form and documenting things that did not seem to relate to my workflow nor seeming to add value drove me crazy. And sometimes I would challenge the powers that be about these situations only to hear a response that stated I had to document this in order to be compliant.
Fortunately, I learned years later that there are a litany of ways to interpret medical device regulations. Most interpretations I have observed are to dictate over-documentation.
I have a different point of view: document the bare minimum to demonstrate compliance and keep document management as simplistic, straightforward as possible. Said another way, as a medical device professional, I should agree that the things I document are value-add events.
Another point of contention relating to document management is internal misalignment of what is expected and required to be documented. And to illustrate this a bit further, I’d like to share a brief story from when I started my consulting practice many years ago.
The engagement involved providing support to help a startup with their QMS. I stepped in to help another consultant, who at the time had responsibilities for quality, regulatory, and clinical. Efforts were ramping up on the product development front and having one person to effectively manage quality, regulatory, and clinical was just too much.
When I got engaged, the QMS had already been drafted. My first task was to familiarize myself with the “established” QMS. I did so by doing an audit of sorts to compare the QMS against FDA 21 CFR Part 820 and ISO 13485. The results of this audit was thumbs up; the QMS met the requirements defined by FDA and ISO.
My next step was to continue with audit-like activities to compare the company’s documentation against the SOPs within the QMS. I learned very quickly that the actual company documentation practices and workflows did not align with the QMS. Or maybe better stated: The QMS SOPs were written in a way that did not match how the resources actually conducted their work.
Sadly, variations of my story happen a lot in the medical device industry. It’s almost as if those responsible for defining QMS and documentation requirements define the criteria in a vacuum and do not include the process owners and stakeholders in the process.
Distinguish between document control myth and reality
There are often unacknowledged assumptions that leave existing document control problems hidden from companies’ view.
These common myths make it easy to believe that the system working against you is actually working well enough.
- Misplaced Desire to Innovate
Myth: Too much focus on document control will stifle innovation.
This simply isn’t true. What slows down innovation more than anything is asking team members to locate misplaced documents or worse, much worse, failing to find a requested document in the middle of an audit. Warning letters stifle innovation more than documentation ever will.
- Misplaced Desire to Save Costs
Myth: Paper and general-purpose tools are the cheapest and easiest solution.
This may be true during the initial phase of using these tools, but the costs will inevitably add up.
Paper systems restrict collaboration and make it difficult to track document versions. That’s troublesome when most companies don’t keep all their staff in one building, especially in the era of coronavirus and remote work. And as your company scales and grows, paper-based approaches are nearly impossible to maintain effectively over time.
General purpose document/file management tools like Dropbox and Google Drive offer some basic tracking features, but don’t comply with FDA 21 CFR Part 11, and come unvalidated. It’s only a matter of time before the costs saved upfront will resurface later in compounding fashion.
- Misplaced Desire to Remain Compliant
Myth: It’s difficult to ensure an eQMS is compliant.
FDA 21 CFR Part 11 requires the meeting of strict standards for implementing and operating an eQMS.
If you’re not using a purpose-built, industry-specific QMS, then customizing, managing, and validating tools like SharePoint will seem like more of a hassle than sticking with paper. With a purpose-built Medical Device QMS, such as Greenlight Guru, the Part 11 compliance guardrails are built-in.
Identifying and correcting these assumptions is the first step toward regaining document control. Once you can see past the lure of legacy systems, you can start working your way out of them.

5 Benefits of Document Management for Medical Device Companies
Once you have an effective document management system, you can leverage it in numerous ways across your organization.
Document control isn’t a single procedure, but rather an ongoing process that extends across the lifecycle of the product, influencing processes at every stage. The better you hone this process, the better you can leverage these five advantages of an effective document management system.
1. Better accessibility to documents
A good document management system makes every single one of your documents, and every single version, quickly and easily accessible. This enables you to inspect and review documentation before signing off on new stages of the product lifecycle. If something seems amiss, you can revert to previous versions and trace any component to identify and correct any issues found.
Good document management streamlines change management so your team is no longer playing ping pong with your change packet. Automatic version control means your documents are always up to date, and old versions are inaccessible to anyone without the right document-level permissions. Said another way, a good document management system should easily and effectively take care of document control.
2. Greater responsiveness from key stakeholders
Good document management creates a more responsive organization. This level of responsiveness can inadvertently provide a huge boost to team morale, as well. Hunting for documents can become contentious. It can lead to finger-pointing and accusations as to who’s responsible, a situation fraught with strife that can plague teams.
Instead, teams should have the tools that allow them to channel that energy through a collective response to solving challenging problems or creating innovative quality designs. In the case of an inspection, a responsive team is able to fulfill every document request immediately, instead of scrambling to surface the necessary information and hoping it will suffice.
A team with good document management can be proactive, not reactive. Rather than expecting to manage any situation through damage control mode, proactive teams stay ahead of regulators and deal with issues before they plant their roots and become systemic.
3. Easier compliance activities
One of the major differences between modern, purpose-built document management and control compared to legacy document control is the availability of system-driven compliance instead of employee-driven compliance.
Employee-driven compliance is carried out entirely by the individuals within an organization who are responsible for creating, following, and finding documents that demonstrate compliance. This can mean long days and burdensome, manual tasks. It’s not fun work, and it’s certainly not rewarding work.
System-driven compliance relies on the document control and quality management system to automatically generate, organize and track documents. When your system drives compliance, document control is preprogrammed to enable compliance with seamless tracking mechanisms, like determining which documents have been executed by whom and when.
Let me share a short example from a Greenlight Guru customer.
The team at PhotoniCare was working on a device that helps pediatricians better diagnose ear infections when they realized their manual documentation system was becoming increasingly burdensome and too unwieldy to ensure future compliance needs.
After implementing Greenlight Guru, the team remembers feeling an immediate difference in terms of the control they had over their growing volume of documents. They were no longer spending countless hours manually assembling their design history file, Greenlight Guru provided one out-of-the-box that was ready to turn into their own.
4. More accountability around business practices
Document control enables your organization to create greater levels of accountability. This benefits not just your teams––in-house and remote––but also your processes.
With the right document management system, you can set up review tracks for each document type. That means that each time someone uses a document, your system indicates the revision history and what changes have been made over time.
This is particularly advantageous to teams whenever there’s a misunderstanding about a process; document control provides a means for accountability that you can use to identify and correct any incongruences. This level of accountability ensures objectivity and clarity from start to finish.
Accountability isn’t a purely punitive measure. When you set up review tracks, workflows become streamlined as a result. Only the person(s) responsible for managing a particular change will be assigned, so there’s a huge time-savings advantage that transpires from this, as well.
5. Improved document security
A good document management system keeps your documents, and the information contained inside, safe and secure. An ad hoc document control system tends to rely on the security practices of outside vendors that don’t share your incentives.
Your email, storage, and messaging providers haven’t necessarily accounted for the specific security parameters necessary to ward off potential threats and to meet the security requirements of the medical device industry.
A cloud-based QMS with a document management system like Greenlight Guru, on the other hand, provides a secure experience tailored to the needs of medical device companies. Users can set document-level permissions, as well as user-based permissions, to ensure you’re providing the appropriate level of document access to the correct resources.

How to regain (document) control within your QMS
The problem is clear. When you’re not in control of your documents, you risk running afoul of ISO and FDA requirements. Moreover, you risk losing your ability to manage quality throughout the lifecycle of your device.
Effective document control AND document management is within your reach.
Good document control enables collaboration and compliance. As different users update and circulate artifacts, everyone can stay engaged in the process and work on the right documents. Version control keeps them organized, and your QMS ensures traceability between moving parts.
This is the vision to work toward.
Right-size your document control system as your company grows
Document control is core to your QMS. But often, we see a QMS is out of sync with the size of the company using it. That results in document control that offers little to no control.
You don’t need your entire QMS ready on day one. You can build your QMS in stages, in tandem with your company’s growth.
That’s why we recommend right-sizing your QMS as you build it. This means molding the size of your QMS to match the size of your company. Right-sizing your QMS ensures you build the quality procedures you need, as you need them.
A sprawling QMS makes it hard to see document control needs amid a multitude of other processes. Quality systems tend to either be too light, using language that’s vague and ambiguous, or too heavy, using language that burdens your team with an overwhelming amount of specifics.
A right-sized QMS gives you the space to prioritize document control. You can focus on a few documentation aspects to set yourself on the right track as you begin to right-size your QMS.
The first is performing an internal audit, which can provide valuable insight into the performance of your internal processes to reveal what’s working and what might require deprecation. The second is conducting a gap analysis of your QMS to understand which procedures must be in place for compliance and how they should function.
When your QMS fits your company, you’ll find it won’t be too unwieldy to follow or so small that it lacks the capacity to manage key documents. When you strike this balance, true quality becomes a natural byproduct.
You can maintain this balance by following a phased approach and build out the rest of your documentation as your company evolves.
Build a documentation strategy to guide your growth
Documenting without a plan is risky. You may end up documenting too little, leaving you vulnerable to announced FDA inspections, or you may end up documenting too much, creating a burdensome sprawl of documents.
The key, similar to right-sizing your QMS, is to determine your needs ahead of time and build your documentation from the ground up.
Think carefully about medical device product realization and the lifecycle of your device. Map out all the types of documents these processes will require you to generate.
Before you actually document a single thing, determine who owns each document, how long each document needs to be kept, and where you’re storing these documents.
From here, you can start building a document strategy that will guide your growth.
Rebuild document management in phases when your company is established
When your company is more established, improving and updating your document management requires a different tack.
We recommend breaking down your document management system into phases and attacking each phase with a well-thought-out plan.
Early-on, it’s relatively easy to take apart your processes and start over.
For midsize to large companies, however, rethinking systems and processes can be daunting. By the time your company is employing 50, 100, or even 200 people, there are all sorts of personnel, job duties, and tools tied up in existing systems.
It’s easy to feel “in debt” to the tools and approaches you’ve already established by continuing to invest in it, rather than consider the possibility that a total divestment and reinvestment may be your best bet. In other words, the short-term costs of rebuilding eclipse the long-term benefits of building a sustainable system for document management.
This can be an intimidating thought for companies because it’s assumed that the rebuilding process needs to happen all at once.
Create a quality plan, break each process into tasks, and schedule each task to be completed within a practical timeline. If you follow this direction deliberately and carefully, rebuilding your document management system will be easier than you initially thought.
Let me share one more Greenlight Guru customer story from Centese.
Centese was managing their design controls and risk management using Excel, updating spreadsheets and manually documenting any changes. This became too burdensome and time-consuming of a task, especially with their sights set on achieving 510(k) clearance from FDA.
Fast forward to now where the entire Centese team finally is able to enjoy value-add activities through the adoption of a purpose-built document management system. Their team can streamline processes and maintain total control over their documentation while also having full traceability of documents and other connected processes within the QMS platform.
Did I mention they also received 510(k) clearance in record time after implementing Greenlight Guru?
Despite the initially daunting prospect of rebuilding a QMS, the Centese team found it surprisingly easy. With the backing of Greenlight Guru and a solid plan, they were able to build a new QMS and reap all the benefits of a better system––all without the strain many companies might expect.

Types of document control systems
Let’s talk about the elephant in the room here: your actual document control systems.
Every company, starting with the smallest startup to the largest enterprise, has a document control system. The real question is whether or not it’s good.

Your tools greatly influence which best practices you can actually adopt. Let’s go over the three document control systems most commonly used by medical device companies.
1. Paper-based and ad hoc document control systems
For decades, medical device manufacturers have relied on paper-based document control systems. This means folders of documents, stacks of binders, and rows of filing cabinets.
Our 2020 State of Medical Device Product Development and Quality Management Report findings showed that over 50% of medical device companies still use paper-based or ad hoc systems.

Paper systems are inherently cumbersome. Early on, when your company is small enough to occupy one room in one office, it seems doable. You can literally pass a document around a table and get everyone’s signature. What could be easier?
As your company grows, a system like this quickly becomes harder. As soon as you involve a third-party vendor or hire remote team members that operate beyond the vicinity of your office walls, your paper system will start to tear.
It doesn’t take long for modern companies to introduce some level of digitization. Rather than pass around a physical document, teams start faxing, printing, scanning, and emailing. Teams might use cloud services but it tends to be in an ad hoc manner without system or procedure.
Despite a layer of technological paint, these slightly better systems inherit many of the same problems exclusively-paper systems have.
It’s as easy to misplace documents and lose track of the most recent version among a multitude of Google Drive folders as it is among a multitude of physical folders.
What may be worse, you’re relying on the security of whichever email hosting tool you and your vendors are using to communicate and transfer confidential information—a regulated process that must be followed by companies for compliance.
Eventually, you’ll need a level of traceability that your ad hoc system can’t implement. You’ll need to access information related to the risk of your device to correlate it across different pieces of quality data, and you’ll be stuck with paper—or digital paper—that can do no such thing. Google Drive can help different team members access the same document, but it won’t prevent users from working on an outdated version.
Paper systems, due to their inherently ad hoc nature, are limited and not built to withstand the complexities involved in overseeing the operations of a medical device.
2. General-purpose document control systems
At first glance, general-purpose tools seem like a solution to the problems that plague document management in paper-based and ad hoc systems.
These tools are designed to help companies manage a variety of quality processes but, because they’re used across industries, they lack many features that medical device manufacturers need.
They don’t come with templated SOPs and workflows suited to the medical device industry, meaning companies must configure all of these details themselves. Importantly, general-purpose systems don’t integrate the regulatory requirements that can make or break a medical device manufacturer.
Validation to 21 CFR Part 11 is an arduous effort the first time around, let alone the repeated efforts that will follow time and time again to ensure compliance with ever-changing regulatory requirements. Real-time access to reliable quality data is difficult, if not impossible.
The inefficiencies of these tools make it nearly impossible to demonstrate closed-loop traceability between your medical device design controls, risk, post-market activities, and other key process documentation.
General-purpose tools may feel like an upgrade over paper systems, but they degrade over time. Since they’re not purpose-built for any one specific industry, they require regular reconfiguration and validation—which means regularly hiring outside firms to contract this work.
In effect, you’re always playing catch-up to a system that was never designed to do what you need it to do. Just when you think you have it right, a new standard or update to a regulation will emerge, and you’ll again have to redirect resources for reconfiguration.
3. Purpose-built document control systems
Purpose-built document control systems are key differentiators.
The best QMS software solutions are purpose-built for the medical device industry that enable document control practices to operate at peak performance. They facilitate the design, development, manufacturing, and distribution of safe and effective medical devices to end users.
Safety guardrails are built-in throughout purpose-built systems to support companies with achieving and maintaining compliance.
Your documents, rather than being a tangent to your core processes, can take a central role in a connected quality ecosystem. With a purpose-built system, you can track and update documents throughout the lifecycle of the product, maintain closed-loop traceability, and integrate all the quality processes, design inputs, and risk data you need.
Greenlight Guru offers the only QMS software purpose-built for medical devices. The software comes out of the box with advanced document management workflow capabilities with Part 11 compliant e-signatures, approval workflows, revision control, and more, to ensure peak performance amongst teams working under one roof or those working remote.

Click here for your free demo of Greenlight Guru’s Advanced Document Management Software
Greenlight Guru comes with a validated OQ/PQ process that covers the required components of Part 11. This is only possible because Greenlight Guru is built on top of a multi-tenant cloud architecture with medical device professionals in mind.
Every time we release a new version of our software, we put it through a battery of tests that reflect the same level of comprehensiveness we recommend to our customers. Updates, enhancements, and new features are automatic due to a cloud-based technology stack.
Some general-purpose QMS tools may offer document control, but there’s only one QMS software that provides closed-loop traceability and enables you to go beyond creating and storing documents to managing documentation for every process throughout the product lifecycle.
A purpose-built system becomes your single source of truth that stays up to date as your company matures. This is important because document control will suffer unless you can manage it via a system that scales with company growth.

How document Control Improves Change Management
Good document control makes it easier to keep track of who has access to what, which version they’re working on, and where the newest version is located. Once you can better manage changes, you can better collaborate within your team and with external stakeholders.
Consider an example: Let’s say you’re ordering a manufactured part for your medical device. When you make this purchase, you always want to have the newest version of your product specifications available.
If you’re using paper or a general-purpose document sharing tool, you have to worry about vendors referencing old versions of documents.
With a general-purpose system, you might share product designs with your supplier via email. If you update any information, the supplier now has an outdated version. You might not even remember who has which version, so you can’t ensure vendors are always working on the most recent versions.
You can tell your team to keep track of constantly-updating documents, but it’s harder to tell, and expect, vendors to do that. As much as you may have vetted these partners, they’re working with numerous companies like yours.
We recommend a risk-based approach to qualifying vendors. Outsourced vendors can save you money, but without due diligence, vendors can also produce products that don’t meet your standards. This might mean more audits, product returns, or product rehauls.
Document control is a key part of your ability to qualify vendors.
Without document control, you could end up ordering parts or tooling that don’t align with current specifications. Your vendor might manufacture parts or products according to an outdated document that never got updated. That could translate to thousands of dollars and weeks’ worth of time wasted due to poor documentation.
A purpose-built QMS and document control system makes this process easy—almost hands-off. Purpose-built systems provide version control and traceability, meaning you can backtrack from newer versions to older versions to trace changes and archive outdated versions.
Your goal is to create a single source of truth. Your document control system should tie together change control with one approval and routing process. It’s possible to build a document ecosystem that involves your team and an extensive array of partners where no one has to worry about working off of a document that has become outdated unbeknownst to them.
Check out our Definite Guide To Change Management For Medical Devices to learn more.

How Document Control Improves Inspections and Audits
Audits seem like they’re all about the technicalities, but they’re just as much about perception. Good document control helps you frame your company and its processes to auditors.
Imagine this scenario: You’re undergoing an audit when the auditor asks for your CAPA documentation.
You remember this corrective action clearly: One of your most incisive engineers detected a flaw in a device that prevented it from functioning as intended under certain circumstances. Your team investigated the issue and identified the corrective action to be taken.
Eventually, your team made you proud: They corrected the flaw and documented everything, and no patient was harmed along the way.
There’s one problem: Where’s the document that recorded it all?
To the auditor, all that work your team did to correct the flaw is immaterial.
If it wasn’t documented, it didn’t happen.
“But it did happen!” you might want to say. You did document it. But similarly, a document lost is just as valuable as a document that was never created.
As much as you may have intended otherwise, poor document control can lead straight to a Form 483 observation or warning letter from FDA.
If you struggle to find the requested documents, inspectors are going to dig deeper because you’ve proven they can’t trust the system you have in place.
When documentation becomes outdated, it’s like whack-a-mole trying to locate and update old versions. With an inspector waiting, your team can spiral, scrambling all day and night to put together the requisite documents. This means stress on your team and, worse, an inspector with too much time on their hands.
Good document control keeps inspectors and auditors busy because you can always pull up the documents they request at a moment's notice.
Every time they ask for a document, your document control system can surface it with the click of a button, and you can hand it over—all the while knowing it’s the most current version that has all the requisite signatures.
The value this offers your team is difficult to quantify. You don’t have to ask your team to spend precious resources finding or updating documents. Those 16-hour crunch days before and during audits are eliminated. Quality professionals spend less time playing traffic cop with signatures and more time upholding quality. It’s transformative.

Optimize your document control with a medical device QMS solution
If it’s not documented, it didn’t happen. That’s the philosophy we have at Greenlight Guru and the philosophy we recommend to all of our medical device customers and the medical device community alike.
When you adopt this same type of mindset with your own documentation, document control no longer becomes a tangent you tack on to the design and development process of your medical device. Instead, documentation becomes an essential process that runs concurrently alongside all of your other processes.
When documentation is so essential to your success as a company, as it is for any company in the medical device industry, good documentation procedures come naturally. Most often, this also means buying instead of building.
Building your own document management system is like cobbling together scaffolds instead of buying a ladder. You know you’re going to have to fix it even as you climb to new heights. The only QMS software that can get you there is from Greenlight Guru. We built our solution to support your team and help you reach new heights alongside your company's growth.
Looking for a document management solution to help you bring safer medical devices to market faster with less risk and ensure success of your products post-market? Click here to take a quick tour of Greenlight Guru's Medical Device QMS software →
Free eBook: Ultimate Guide to Document Control for Medical Device Companies

%20Ultimate%20Guide%20to%20Document%20Control%20for%20Medical%20Device%20Companies.png?width=180&name=(cover)%20Ultimate%20Guide%20to%20Document%20Control%20for%20Medical%20Device%20Companies.png)
%20Ultimate%20Guide%20to%20Document%20Control%20for%20Medical%20Device%20Companies.png?width=250&name=(cover)%20Ultimate%20Guide%20to%20Document%20Control%20for%20Medical%20Device%20Companies.png)







