
It’s easy to get a bit confused when you consider the records that are required for your medical device.
The industry is full of acronyms, with many of them sounding very similar. If you’re involved with medical device development, it’s important that you’re able to grasp the difference and be compliant with quality management regulatory requirements.
Three such device records that are commonly mixed up are: Design History File (DHF), Device Master Record (DMR) and Device History Record (DHR). They all sound very similar, right? These particular terms often create a lot of confusion because they share many similarities, but I assure you, each serves an important role in your design and development process.
For medical device professionals, it's critical to understand the purpose of each of these terms, as well as their differences. Let’s take a closer look.
Free Download: Click here to get our quick guide to device records and when to use them.
Design History File (DHF)
You’ll find the Design History File referenced by the FDA in 21 CFR Part 820:30. In fact, your DHF has everything to do with Design Controls, because it is the last step in which you compile the documents from your design and development process.
As the FDA states: “Each manufacturer shall establish and maintain a DHF for each type of device. The DHF shall contain or reference the records necessary to demonstrate that the design was developed in accordance with the approved design plans and the requirements of this part.”
Direct mention of the DHF is made in the updated ISO 13485:2016 too, whereas it was not previously referred to specifically. The ISO standard now specifies the need to establish “design and development files.”
In the beginning of the design and development phase, you're working on your device's Design Controls. The overall purpose of this process is to demonstrate that your medical device is safe and effective, and that it meets the indications for use.
As part of this process, you need to record: Design and Development Planning, Intended Use and Indications for Use, User Needs, Design Inputs, Design Verification, Design Validation, and Design Reviews and Design Outputs. Industry best practice states that you should keep a traceability matrix showing the linkages between key elements too. ALL of these need to be contained within your Design History File.
There are some key points you should take into account for your DHF:
- It’s much easier to start sooner rather than later. A key mistake is scrambling to put the DHF together in time for 510(k) submission, or an FDA inspection. Your DHF is a central source of a lot of things that medical device regulators care about, so expect it to receive some scrutiny, and put an appropriate amount of time into keeping it updated.
- It is your responsibility to prove traceability and linkages between the DHF elements. Make it easy for the FDA by creating a robust matrix.
- Your DHF should be well-organized and accessible to everyone on your project team.
- Your DHF is HUGE. You’ve seen all the parts that go into it - if you’re recording manually, this can easily run into multiple binders of information. An electronic, cloud-based system can be a great solution for keeping information safely stored, easily updated and accessible to those who need it.
- Keep your DHF separate from your project file. The last thing you will want to do after the development process is to go back and clean up all of that paperwork!
Device Master Record (DMR)
Your Device Master Record (DMR) contains everything you need to know in order to build and test your device. You will find the FDA’s regulations for the DMR under 21 CFR Part 820:181.
You could look at the DMR as a recipe for building your device. It contains all of the specifications to build the device from start to finish. It isn’t concerned with design history (unlike your DHF), but instead is concerned with the current information and specifications needed to produce your device from start to finish.
FDA’s requirements state:
Each manufacturer shall maintain device master records (DMR's). Each manufacturer shall ensure that each DMR is prepared and approved in accordance with 820.40. The DMR for each type of device shall include, or refer to the location of, the following information: (a) Device specifications including appropriate drawings, composition, formulation, component specifications, and software specifications; (b) Production process specifications including the appropriate equipment specifications, production methods, production procedures, and production environment specifications; (c) Quality assurance procedures and specifications including acceptance criteria and the quality assurance equipment to be used; (d) Packaging and labeling specifications, including methods and processes used; and(e) Installation, maintenance, and servicing procedures and methods.
Let's consider another example for how DMR works. One of your product engineers designs a specific part that forms an attachment on your medical device. They draw the design, and that becomes part of the Design Outputs section of your DHF. The difference with the DMR is that it would also include specific instructions for manufacturing the part. This might include production methods and anything else that is important to produce the part as designed.
Your DMR should include the following, according to the FDA:
- Device specifications including appropriate drawings, composition, formulation, component specifications, and software specifications;
- Production process specifications including the appropriate equipment specifications, production methods, production procedures, and production environment specifications;
- Quality assurance procedures and specifications including acceptance criteria and the quality assurance equipment to be used;
- Packaging and labeling specifications, including methods and processes used; and
- Installation, maintenance, and servicing procedures and methods.
Another key difference to understand is that your DHF is produced during design and development, whereas your DMR is established during the transition from development into manufacturing. Your DHF still plays an important role, particularly for your Design Outputs. These can form the basis that kicks off your DMR.
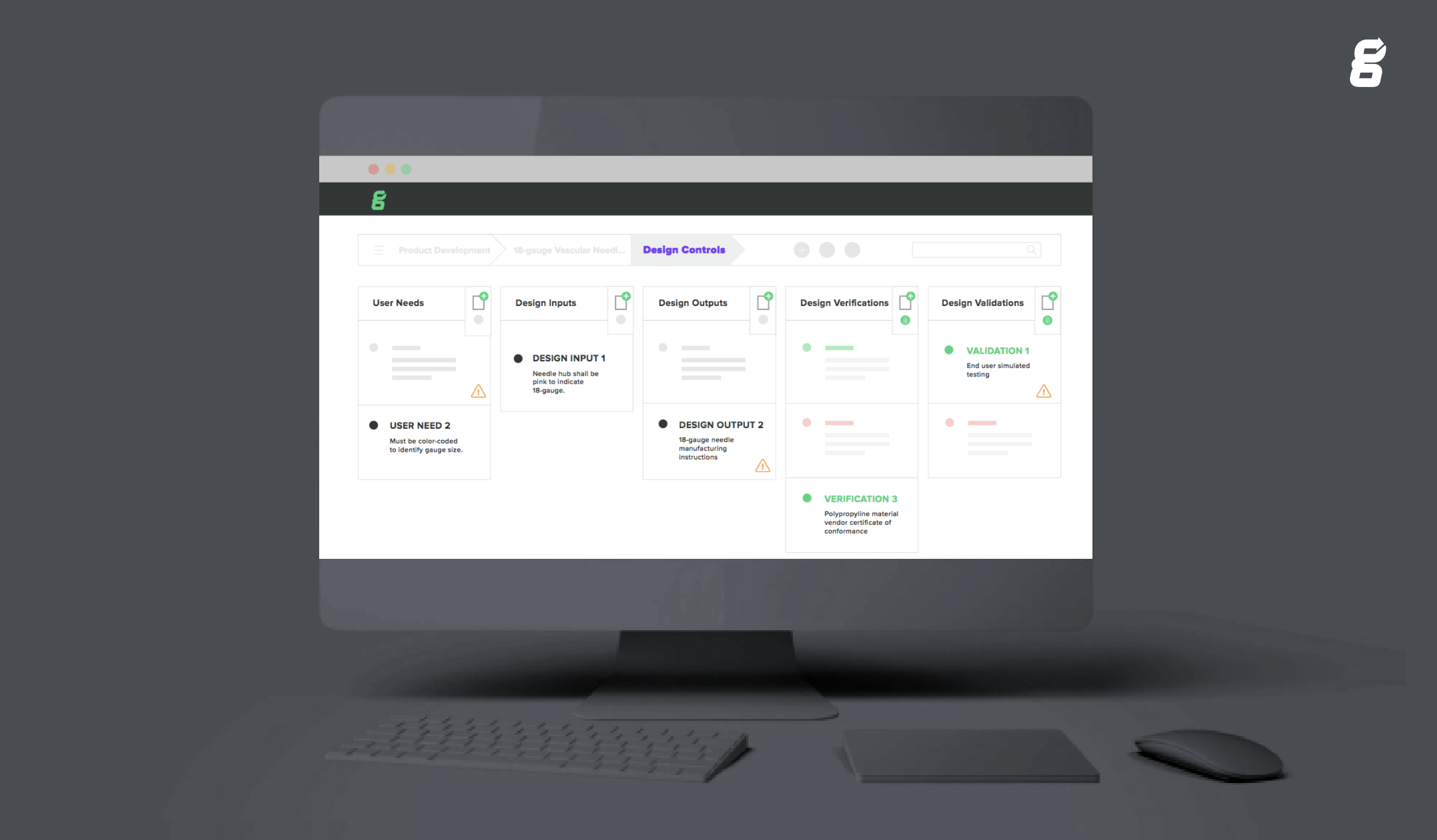
Learn how Greenlight Guru's Multi-level Design Control software simplifies device record keeping
Device History Record (DHR)
The Device History Record (DHR) is actually the next document in line if you look at these in chronological order. It is one of the last steps in the compliance process for medical devices.
Your DHR contains all documentation related to manufacturing and tracking your device. It demonstrates that you manufactured the device according to the information in your DMR.
You can find the FDA’s requirements under 21 CFR Part 820:184. This is under subpart M of Part 820, which deals with record-keeping (whether it be paper-based or electronic). As for what should be contained within your DHR, here’s what the FDA says:
Each manufacturer shall maintain device history records (DHR's). Each manufacturer shall establish and maintain procedures to ensure that DHR's for each batch, lot, or unit are maintained to demonstrate that the device is manufactured in accordance with the DMR and the requirements of this part.
The DHR shall include, or refer to the location of, the following information:
- The dates of manufacture;
- The quantity manufactured;
- The quantity released for distribution;
- The acceptance records which demonstrate the device is manufactured in accordance with the DMR;
- The primary identification label and labeling used for each production unit; and
- Any unique device identifier (UDI) or universal product code (UPC), and any other device identification(s) and control number(s) used.
The DHR is different from other quality processes because it is applied to each individual batch of devices. In other words, each new batch produced should be tested to ensure that the products meet the specifications laid out in the DMR.
Your DHR is also useful as a tool for facilitating CAPA investigations and process improvements. You can determine UDIs or UPCs associated with a complaint and drill down on a root cause for individual batches. You can also trace non-conformance issues to inform process improvements.
The DHR is your overall history of the device, much like the DHF is the overall history of design.
Free Download: Click here to get our quick guide to device records and when to use them.
Final thoughts
DHF. DMR. DHR. At first glance these acronyms might have seemed similar, but now you have a greater understanding of their differences and how they function in the design control process.
If you still need some help distinguishing these tricky terms, try these word associations: you must have a File for your Design and a Record of your Device. You can also think of it in chronological order, starting with the Design History File in design and development, then the Device Master Record during the production phase and finally the Device History Record once the product is in the manufacturing stage.
These key terms and their associated processes are built into automated workflows of Greenlight Guru's Quality Management Software. Our cloud-based platform hosts a library of forms, templates, and procedures to streamline all documentation in a secure, single location. Designed specifically for medical device companies, Greenlight Guru is designed specifically for medical device companies in order to simplify regulatory compliance, specifically the record keeping requirements mandated in 21 CFR Part 820.



