_%20Substantial%20Equivalence%20Through%20Performance%20Criteria.png?width=2400&name=Abbreviated%20510(k)_%20Substantial%20Equivalence%20Through%20Performance%20Criteria.png)
A recent draft guidance document was published by FDA detailing the expansion of the Abbreviated 510(k) program. Establishing substantial equivalence through performance criteria is the most notable update to this guidance. With the comment period now closed, the final version of this guidance is officially underway.
Medical device manufacturers may choose to submit an Abbreviated 510(k) when there are FDA-established performance criteria. The Abbreviated 510(k) is a pathway that has been around for a little while, although records show that the FDA receives less than 100 applications via the Abbreviated form per year.
What should device manufacturers know about the Abbreviated 510(k) and the proposed updates?
Types of Premarket Notification 510(k) Submissions
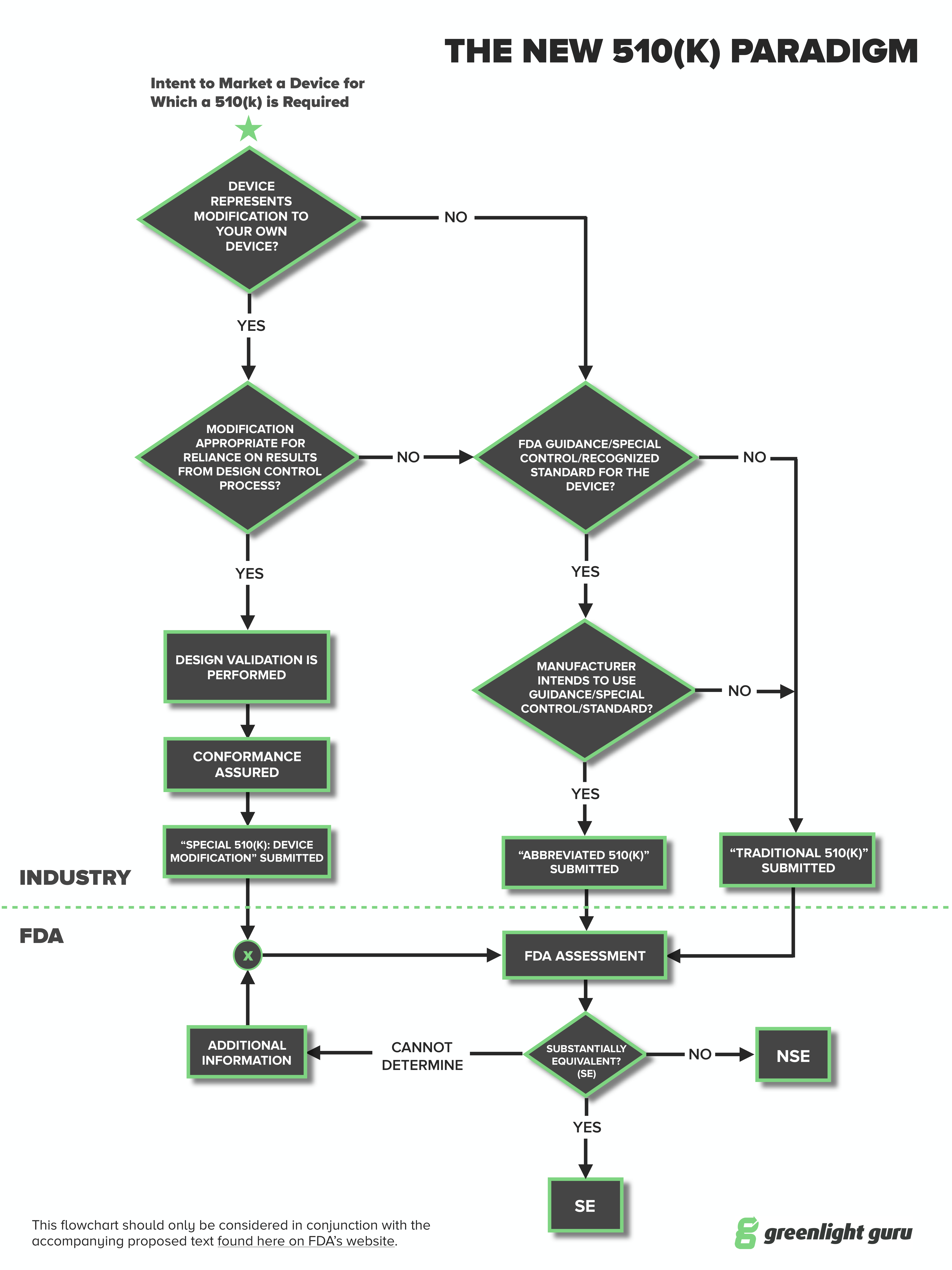
The Abbreviated 510(k) is one of three types of premarket notification 510(k) submissions, with the other two being Traditional and Special. Here’s how they compare:
- Traditional - The most commonly used. The essence of this is to show substantial equivalence with an existing or predicate device.
- Special - This is commonly used when you have a device already on the market and want to make a change or modification to it. The change may be labeling, design or manufacturing technique and the Special 510(k) is to notify the FDA of the change. The predicate is your existing device.
- Abbreviated - You have a “paper comparison.” That is, you show substantial equivalence to a recognized standard, special control or guidance using a declaration of conformity.
At the moment, more than 80% of 510(k) submissions are the traditional type.
An “Abbreviated” Pathway doesn’t mean less work
When people look at the term “abbreviated,” they often think that the word implies the process will be less work or perhaps have a shorter review time. Don’t be lured in by the word - it really is no less work.
This pathway does not eliminate the need to demonstrate substantial equivalence. It does not reduce the amount of work. It is not a means to cut corners. This is simply another potentially viable means to help ensure regulatory options and hopefully, a means to provide additional clarity.
Sometimes the review time is greater than a Traditional 510(k). In fact, you can often safely double the review time of the Traditional for the Abbreviated 510(k). The only version that has a shorter review time is the Special 510(k). The average time for these is roughly 50% of the Traditional 510(k) time.
In terms of the format of the Abbreviated 510(k), when applicable, there will be specific performance criteria and, in some cases, corresponding test methods. This can reduce uncertainty about what has to be measured, but it remains to be seen as to whether there will be any less testing compared to a Traditional 510(k). It’s possible that after the review period, criteria for some types of devices may be more demanding with the Abbreviated 510(k) route.
What goes into an Abbreviated 510(k) Submission?
Overall the format is very similar to other types of 510(k). You might even say the format is substantially equivalent to the Traditional 510(k). You’ll find it is largely similar to what you expect from the Traditional 510(k).
The most important difference is the substantial equivalence with a guidance, standard or special control. This is a “paper comparison,” rather than a comparison with a specific device.
The process is no simpler for medical device manufacturers from a product development perspective as you’re still demonstrating substantial equivalence, just with a different methodology. According to FDA,
The Abbreviated 510(k) relies on the use of guidance documents, special controls, and recognized standards [...] Device manufacturers may choose to submit an Abbreviated 510(k) when:
1. a guidance document exists,
2. a special control has been established, or
3. FDA has recognized a relevant consensus standard.
In an Abbreviated 510(k) submission, manufacturers elect to provide summary reports on the use of guidance documents and/or special controls, or declarations of conformity to recognized standards, to expedite the review of a submission.
This is great when I look back at many 510(k) submissions I’ve been part of. Sometimes making that substantial equivalence argument against a predicate device can be very challenging. The idea of using a standard is appealing in these cases.
In terms of meeting the standards, the FDA states that it relies on a declaration of conformity by the submitter without the submission of actual data, although a summary of the data may be needed. Of course, the FDA can request this data at any time, so while not having to include it in your Abbreviated 510(k) submission might be less paperwork, it doesn’t take away any of the work you have to put in behind the scenes.
In terms of the content, it’s important to show why the consensus standards are applicable even though the regulation doesn’t require it. This can include why you didn’t use certain standards as a comparison. It’s entirely up to you as the manufacturer to provide the FDA with enough information to explain your decision. Be thorough with your explanation.
Don’t just explain what you do, explain what you’re not doing and why you’re not doing it.
Factoring in The regulatory environment
There's a big push for device makers to take the “least burdensome” approach possible in terms of a pathway to bring their device to market. However, in the case of the Abbreviated 510(k) pathway, it’s really semantics in terms of the work involved. It’s effectively the same amount of work, but with standards as a comparison instead of a device.
In the European Union (EU), medical device regulations (MDR) tend to be trending the opposite way, with moves away from “paper comparisons.” The new EU MDR that will take effect in 2020 direct manufacturers to do comparisons with predicate devices. This has been generating a lot of angst, particularly among people who are not sure how to get hold of a competitor device or the device information.
However, issues such as these sit firmly in the court of the manufacturer, not the regulator. It is always going to be up to the developer to do their homework. It doesn’t matter if you’re doing business in the US or the EU, the engineering best practices remain the same.
Weigh your options to Determine Best Pathway for your Company
If you’re developing a device and heading down the path to market, it’s a good idea to assess all of the options you have for 510(k) and weigh out their relative merits and drawbacks. Given your company's unique circumstances, technology, business objectives etc., find the pathway that best suits your situation.
Besides that, getting in front of the FDA early and starting that dialogue is a good idea. This could be through the pre-submission process, or even through making contact with them early on. It’s important to sell the FDA on your device early, as submissions that are seen for the first time at the 510(k) point are often a red flag.
It’s our job to prove our assertions. Why is your device substantially equivalent? Remember that with the Abbreviated 510(k), you still need to meet this standard. In fact, the Abbreviated 510(k) RTA (Refuse to Accept) checklist is actually longer than the Traditional 510(k) checklist!
It will be interesting to see how the Abbreviated 510(k) may or may not change after the review period. In any case, it’s up to you as the manufacturer to have done your homework. FDA offers many resources available to the public, including:
Final Thoughts
One of the best ways to supplement all of your hard work along the way is by utilizing a quality management system designed to expedite and simplify your go to market efforts.
At Greenlight Guru, it’s our goal to enhance your medical device product development efforts and streamline your processes through our secure, cloud-based software platform. With our medical device specific solution, companies can electronically review and approve design and development activities at multivariate stages throughout the product lifecycle. Thousands of medical device professionals across the globe are benefitting from Greenlight Guru’s purpose-built QMS Software to bring safer devices to market faster, while reducing risk.




