
As a medical device startup, there are many things you are juggling and balancing all day, every day to advance your product closer and closer to that next critical milestone.
And one of those things you should be doing early on is implementing a bit of infrastructure based on FDA / ISO regulations and requirements.
Doing so ensures that your efforts result in the necessary documentation and records to support regulatory submissions and eventual commercialization of your medical device.
To help, I’ve put together this quick list for you of 15 items your medical device startup will need to address.

#1: Establish Design Controls / Design & Development Procedures
Refer to FDA 21 CFR 820.30.
Create a procedure that describes your company’s design and development process.
Also if you’d like more information on this topic, I wrote about it extensively in the Ultimate Guide to Design Controls for Medical Device Startups.
#2: Define and Document Product User Needs
Define and document Intended Use / Indications for Use.
User needs to describe how the product is going to be used.
User needs help to establish the framework for product design.
Intended use describes the clinical issue your product addresses.
Indications for Use pertain to clinical applications use, environment, and end user.
#3: Establish Regulatory Strategy
Identify applicable regulatory product codes and product classification (based on intended use / indications for use) for all desired markets.
This will help determine the type of regulatory submissions which will eventually be required.
It is important to identify desired markets and determine the regulatory classification for your product in each of these markets.
Here are some links to a few of the more common regulatory markets to help you determine this:
- U.S. FDA Product Classification
- EU Medical Device Directive Product Classification
- Health Canada (CMDR) Product Classification
- Australia (TGA) Product Classification
#4: Define and Document Design and Development Plan
Identify major milestones and define responsibilities.
Establish and maintain plans that describe or reference the design and development activities and define responsibility for implementation.
The plans shall identify and describe the interfaces with different groups or activities that provide, or result in, input to the design and development process.
#5: Define and Document Design Inputs
A Design Input describes physical, functional, performance, and all other product requirements.
Design Inputs are expressed in a way that is measurable and objective.
#6: Establish Design History File (DHF)
DHF contains all Design Controls, including User Needs, Design & Development Plan, and Design Inputs (and all the rest).
DHF should be up to date and organized.
#7: Establish Risk Management Procedure(s)
Refer to ISO 14971:2007 (or EN 14971:2012 for Europe).
Establish Risk Management Procedure(s) with ISO 14971 as the basis.
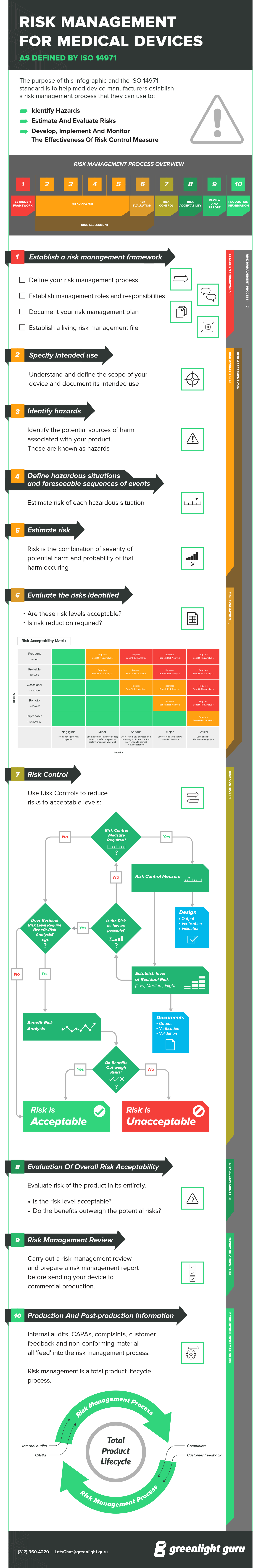
ISO 14971 defines medical device risk management expectations.
I also wrote on this topic extensively in The Definitive Guide to ISO 14971 Risk Management for Medical Devices.
#8: Define and Document Risk Management Plan
The Risk management plan describes timing and type of risk management activities throughout design and development.
#9: Identify and Document Product Hazards
Refer to ISO 14971.
Hazards are a potential source of harm.
Knowing the hazards and hazardous situations pertaining to the medical device you are developing will ensure a safer product.
#10: Begin Documenting Risk Assessment
Risk assessment involves identifying potential harm, severity of that harm, and the likelihood of occurrence.
#11: Establish Risk Management File
The Risk Management File contains all Risk Management documentation.
The Risk Management File is “living” — meaning it gets updated throughout the entire product life cycle.
#12: Establish Supplier Control Procedure
Refer to FDA 21 CFR 820.50.
Formalizing supplier selection and defining evaluation and monitoring criteria will help you choose suppliers wisely.
#13: Create an Approved Supplier List
Determine suppliers and products / services those suppliers are to provide.
Capture your list of supplier on an Approved Supplier List (ASL).
#14: Maintain Supplier Files
Send supplier surveys / evaluations to suppliers to complete and return.
Maintain supplier files.
Your company relies on third-party suppliers which need to be identified, qualified, evaluated, and monitored.
#15: Establish Document Control / Record Management Procedures
Refer to FDA 21 CFR 820.40 and 180, 820.186.
Throughout design and development you need to generate documentation and records and establish a system for organizing and maintain these items.
Additional Information to Consider
Quality System
FDA requires that medical device companies establish a Quality System (QS) compliant with 21 CFR part 820 regulations.
A complete QS is required before going to market.
However, for a startup medical device company still in development, Creo Quality advises that you “bootstrap” your Quality System—or build it as you need it.
As you progress through product development and approach preclinical use, clinical use, and any regulatory filings, additional QS procedures will need to be developed.
And if you have interest in going outside the U.S. build the QS with FDA and ISO 13485 in mind.
Product Development
There are two major aspects of medical device product development which FDA and regulatory agencies are concerned: Design Controls and Risk Management.
As mentioned in the Quality System section, Design Controls are covered in FDA 21 CFR 820.30 and Risk Management addressed in ISO 14971.
FDA also has a Design Controls guidance document, which is worth reviewing.
You should also get a copy of ISO 14971:2007. (NOTE: In EU, ISO 14971:2012 is accepted. The only difference in the versions pertains to a couple of the annexes.)
The most important documentation during medical device product development includes Design Controls and Risk Management.
Design Controls documentation is to be maintained in a Design History File (DHF).
Risk Management documentation is to be maintained in a Risk Management File.

Greenlight Guru Value Calculator

.png?width=1199&name=ROI_Slide-In-02%20(1).png)
%2015%20Items%20for%20Medical%20Device%20Startups%20to%20Address.png?width=250&height=324&name=(cover)%2015%20Items%20for%20Medical%20Device%20Startups%20to%20Address.png)



