See Your ROI with Greenlight Guru in Seconds


Experience the #1 QMS software for medical device companies first-hand. Click through an interactive demo.

Greenlight Guru provides medical device companies with industry-leading software to bring life-changing products to patients faster, more efficiently, and with less risk.

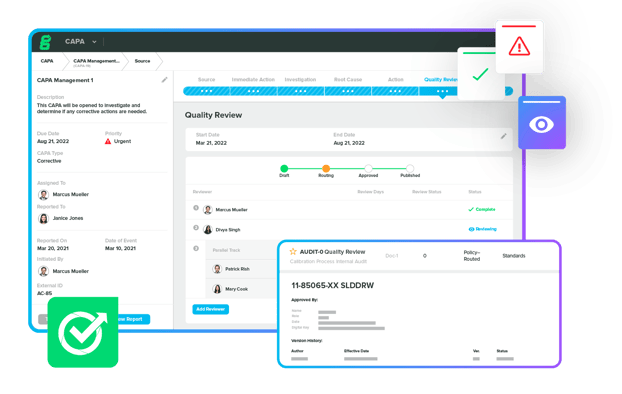
Upgrade your paper-based or generic QMS to a modern, cloud-based solution that enables you to ensure compliance, simplify audit prep, track quality events, and more.
Our QMS software helps you establish a single source of truth while scaling quality throughout your business.
Explore Greenlight Guru Quality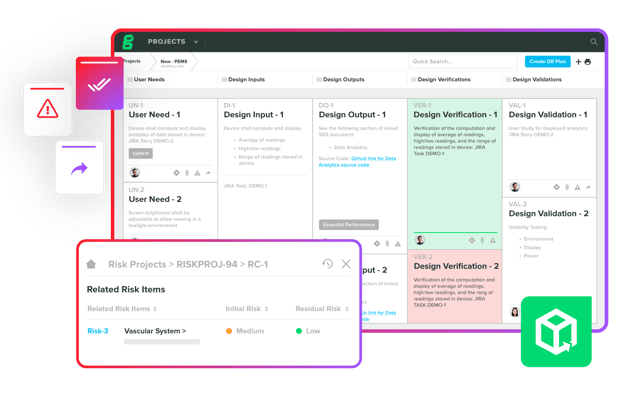
Streamline device documentation, accurately assess risk, and achieve traceability while you iterate through design and development.
Leverage collaborative workflows and automated compliance to accelerate timelines with greater efficiency.
Discover Greenlight Guru Product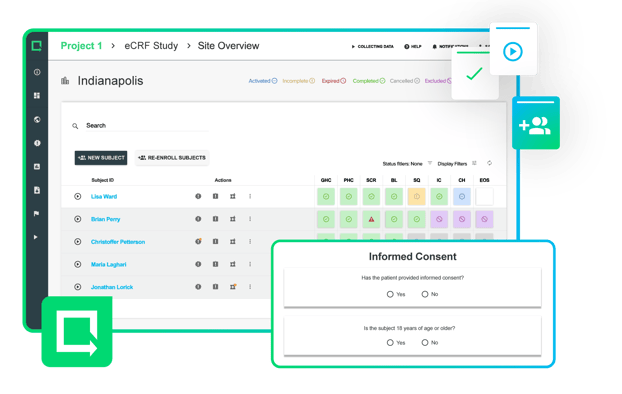
Collect and manage all clinical evidence, safety, and performance data within one versatile digital toolbox.
Modernize your process to cut weeks out of the clinical testing process and bring safer products to market.
See Greenlight Guru Clinical
Greenlight Guru offers much more than software. Benefit from 1:1 human support covering regulations, risk reduction, software guidance, and business roadblocks.
Harness our 500+ years of combined industry experience to confidently meet your objectives at any stage of your journey.
Explore Guru Services


















_Leader_Mid-Market_EMEA_Leader.png)

_Leader_Americas_Leader.png)
_BestRelationship_Total.png)
_Leader_Europe_Leader.png)











