
If you would like to place a medical device on the United Kingdom (UK) marketplace, then changes in the regulatory landscape resulting from Brexit are relevant to you.
The Medicines and Healthcare products Regulatory Agency (MHRA) is an executive authority in the UK that is responsible for governing its healthcare products, such as medicines, medical devices and blood components for transfusion.
MHRA has recently published new guidance on rules that will govern the regulation of medical devices. For some changes, there is a grace period, but others are in effect immediately.
Here’s what you need to know about the post-Brexit medical device landscape and its regulatory gatekeeper MHRA for marketing a medical device in the UK:
UK medical device regulatory landscape post-Brexit
Medical devices entering the UK market, specifically England, Scotland and Wales, will have to follow applicable MHRA UK guidelines. This new rule went into effect January 1, 2021 following the Brexit transition period that ended the day before.
During the Brexit transition period, EU medical device legislation continued to apply in the UK and any EU legislation passed before the transition deadline was automatically incorporated into MHRA requirements.
Make note that the new MDR and IVDR are exclusive to the EU marketplace and do not automatically apply in the UK. One exception is Northern Ireland, which maintains a special status. Medical devices entering Northern Ireland must register with MHRA UK, but follow the EU market requirements of either MDR and IVDR. A CE Mark will remain a requirement for market entry.
From January 1, 2021, the new UKCA (UK Conformity Standard) enters a transitional period for devices entering the UK market. This means that you don’t immediately need the UKCA mark, but you will need to transition eventually.
CE marked devices can be placed on the Great Britain market through June 30, 2030, at the latest, depending on device type and classification.
How is MHRA regulating medical devices in the UK market?
Any manufacturers of medical devices wishing to enter the UK market must register with MHRA. Manufacturers should follow the UK guidance regulating medical devices and must register with MHRA to legally enter both the UK and Northern Ireland markets.
It’s important to note that different types of devices are subject to different requirements, similar to many other regulatory landscapes around the world.
Responsible person
For all manufacturers based outside of the UK, you must appoint a “UK Responsible Person” to register and act on your behalf. Note that for Northern Ireland, you will also need an Authorized Representative based in the EU, if you don’t already have one.
A UK Responsible Person acts on your behalf to carry out certain obligations. Their responsibilities follow the guidelines laid out in the UK MDR (2002). Responsibilities include responding to MHRA and providing the requested information. They must ensure that technical documentation is drawn up and that appropriate conformity assessments have been completed.
The UK Responsible Person will also have a critical role to play in the post-market surveillance of the device. They will need to work with the manufacturer and the MHRA to implement corrective and preventive action (CAPA) that may occur as a result of any complaints or safety issues.
MHRA states the manufacturer responsibilities under postmarket surveillance:
Once a medical device has been placed on the UK market, the manufacturer is required to submit vigilance reports to the MHRA when certain incidents occur in the UK that involves their device. They must also take appropriate safety action when required. The manufacturer must ensure their device meets appropriate standards of safety and performance for as long as it is in use.
MHRA has published a guidance document for manufacturer vigilance in postmarket surveillance. One thing that is clear is how important your QMS will be for documenting and collating your reports and actions. You need to be able to show evidence of how you are meeting postmarket surveillance requirements and, if necessary, taking any FSCAs (Field Safety Corrective Actions).
Recognition of UKCA Mark
The UKCA mark will only be recognized in Great Britain. Existing UK Notified Bodies have become “Approved Bodies” and can conduct the UKCA mark process. They can no longer issue CE Mark.
Additionally, the UKCA mark is not recognized in the EU, EEA or Northern Ireland markets. Here’s a relevant excerpt from the MHRA guidance:
For the purposes of the Great Britain market, UK Approved Bodies can conduct conformity assessments in relation to the UKCA mark, for medical devices, active implantable medical devices and in vitro diagnostic medical devices under Parts II, III, and IV of the UK MDR 2002 (as amended). UK Approved Bodies are not able to conduct conformity assessments in relation to the CE marking other than for the purposes of the “CE UKNI” marking, which is valid in Northern Ireland.
MHRA changes planned for medical devices UK market
Changes to MHRA UK requirements for medical devices are planned, specifically:
-
CE marked medical devices compliant with EU MDR can be placed on the Great Britain market up until the sooner of expiry of certificate or June 30, 2030.
-
CE marked medical devices with certificates issued under current Medical Device Directives (MDD, AIMDD and IVDD) will remain valid up until the sooner of expiry of certificate or June 30, 2028.
-
After June 30, 2030, the UKCA (UK Conformity Standard) will be compulsory for all devices entering the UK market.
-
In terms of labeling, all devices on the UK market must have the UKCA label on them after June 30, 2030. If you already have a valid CE marking on your device, you are not required to re-label the device with a UKCA mark until after June 30, 2030. It will be acceptable for a product to be labeled with both UKCA and the CE Mark prior to, and after that date.
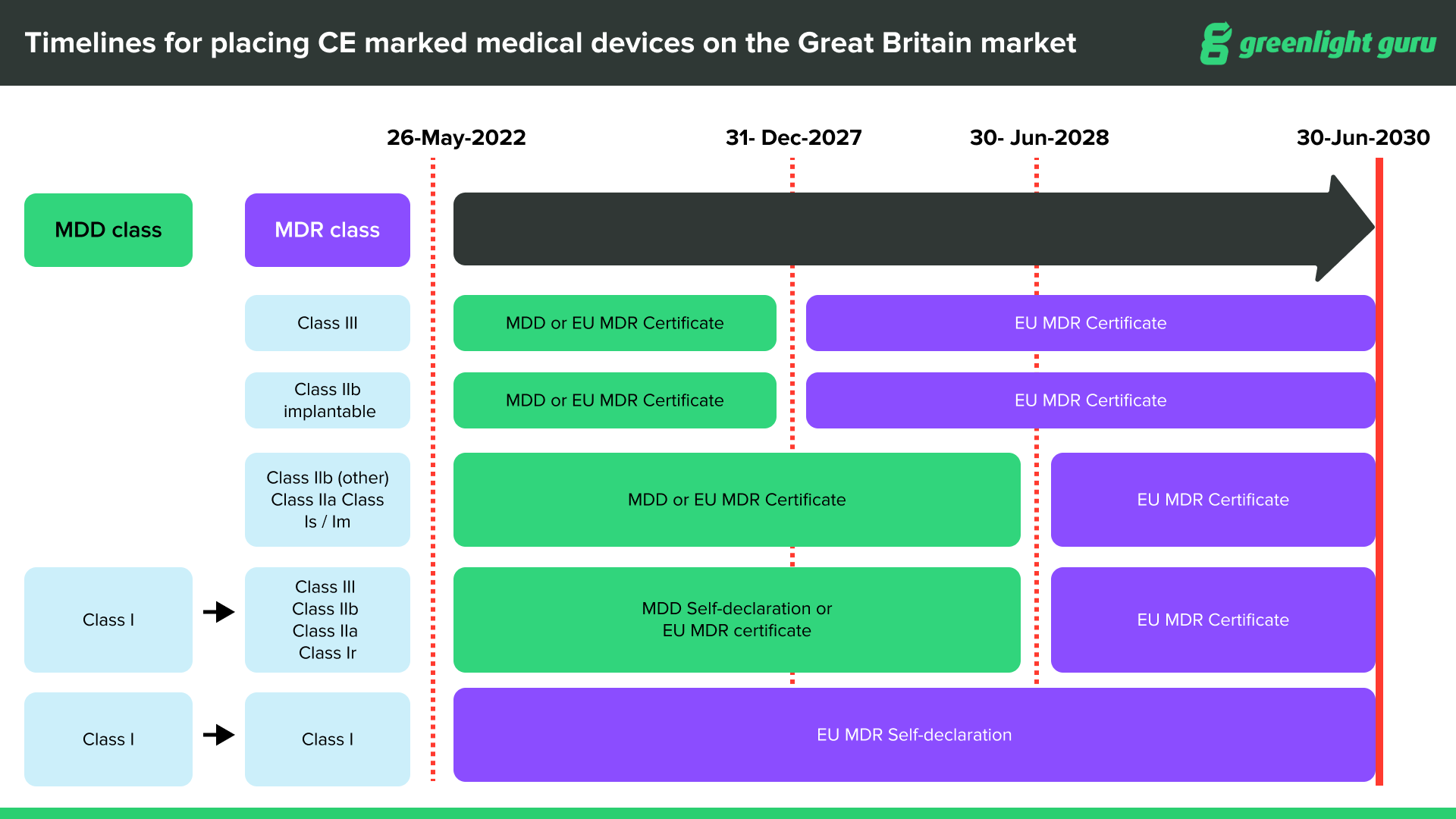
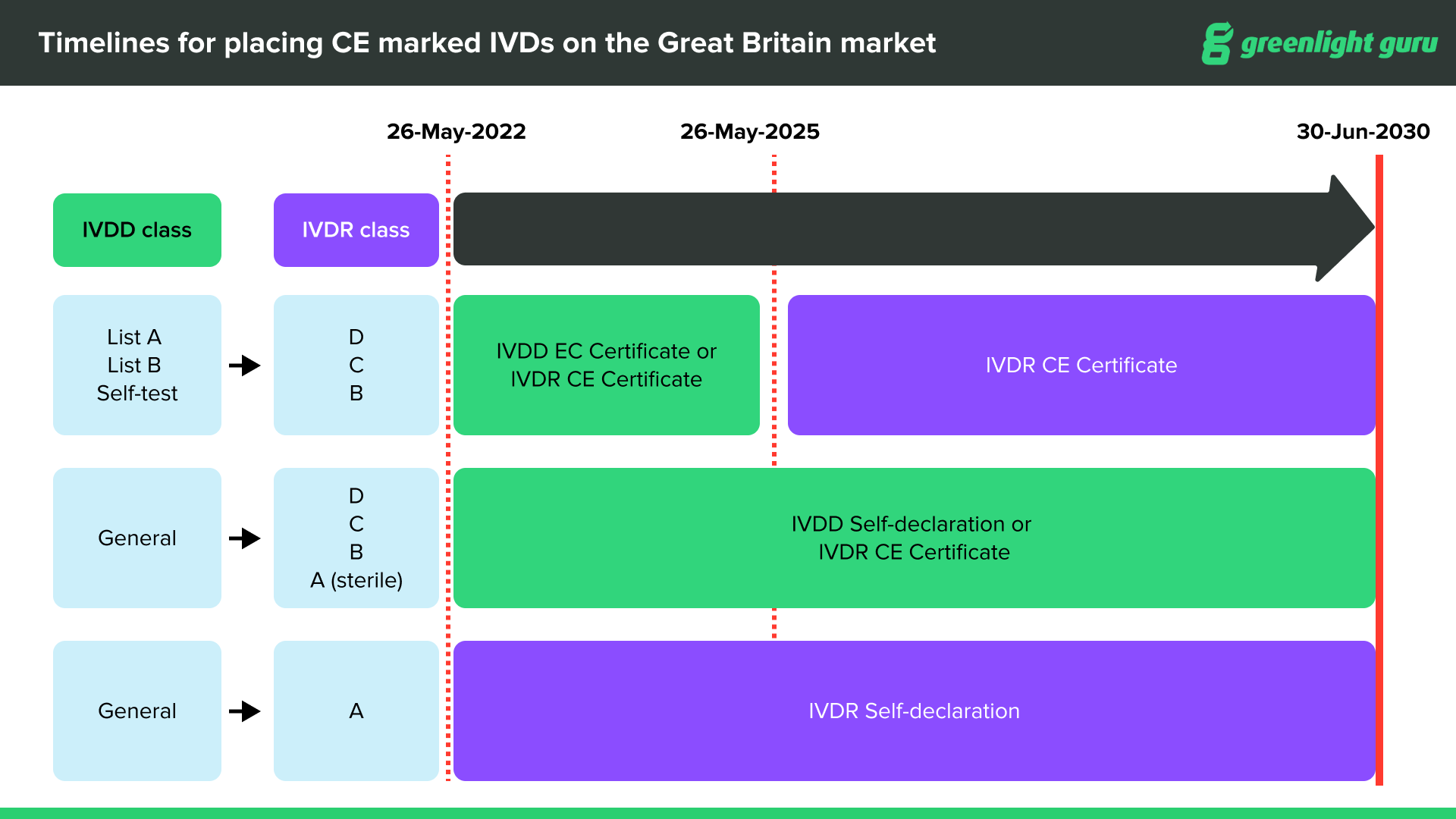
Source: Medicines & Healthcare products Regulatory Agency
Simplify compliance with MHRA requirements to sell your medical device in UK market
If there’s one thing that’s critical to successfully meeting MHRA requirements for medical devices, it’s having a comprehensive, up-to-date QMS that can help you comply with medical device requirements of MHRA UK.
Your QMS should house all of your proof of compliance and importantly, should be as simple as possible for you to use. Greenlight Guru's QMS software—built specifically for medical device companies like yours—will help you to keep all quality system data in order, demonstrate closed-loop traceability, and simplify compliance.
Greenlight Guru is the only QMS solution built by medical device professionals for medical device professionals. Get your free demo today.



