
What should the medical device industry expect in 2017?
Great question.
I’d like to share some of my insights and observations for the upcoming year based on some of the data and trends I’ve noticed in recent weeks and months.
1. A New FDA Commissioner in 2017
A new president often results in a new FDA commissioner. The word on the street suggests that the current FDA Commissioner, Dr. Robert Califf, will likely resign his post after Donald Trump takes office.
Again, this is not all that unusual when a new president takes office.
Prediction: Expect a new FDA Commissioner in 2017.
There are currently a couple names floating around as possible candidates for the new FDA commissioner post. Interestingly, the two names floating around are both involved with venture capital investment.
One of the names rumored to fill the post is Jim O’Neill. Mr. O’Neill is currently Managing Partner with Mithril Capital Management. Interestingly, O’Neill is not a medical doctor, and while not technically a pre-requisite for FDA commissioner, it is often the case.
The possibility of O’Neill as FDA Commissioner is a bit interesting and likely would have a large influence on pharmaceuticals. In fact, O’Neill has stated that drugs should be allowed on the market before their efficacy has been established, as long as they are proven to be safe.
The other name currently being thrown around is Dr. Scott Gottlieb. Dr. Gottlieb is currently a venture capitalist with New Enterprise Associates. Gottlieb would also have a likely influence on pharma.
Regardless, Gottlieb seems to be a bet of a better fit, considering his M.D. background and prior experience as deputy commissioner of FDA.
Califf has only been on the job since February 2016 and his imprint on FDA is too new to have much of a lasting impression.
Califf has been identified as a “big data” advocate. It’s not clear if either O’Neill or Gottlieb will endorse that position.
Regardless, I suspect that either O’Neill or Gottlieb (or another unnamed candidate) will be feeling pressure to get new products--drugs and devices--cleared more quickly.
Verdict: I predict that a new FDA commissioner will bode well for the medical device industry.
2. CAPA a continued area of concern
CAPA is a big, big focus during FDA inspections as well as ISO audits. This has been the case for quite some time.
Why?
Many view the CAPA process as the most critical process. CAPA is the process within a company that provides oversight and visibility into all the ways to improve and fix issues.
Prediction: CAPA will continue to be a hot topic during FDA inspections and ISO audits in 2017.
How a company embraces CAPA can be a make or break scenario. In some cases, companies over use CAPA. Meaning that they realize how important the CAPA process is in the grand scheme of things. So much so, that CAPA is used to capture many things. Maybe too many things.
Other companies do not leverage CAPA enough. CAPA is sometimes an afterthought.
In both situations, CAPA is not being used as intended. CAPA is intended to be a gateway to identifying opportunities for improvement, opportunities to prevent issues, and/or ways to correct problems.
Know that CAPA will be a key part to any FDA inspection and ISO audit. Here is a link to data from FDA’s FY2016 regarding 483 observations. Note the number of citations logged under FDA 21 CFR 820.100.
3. ISO 13485:2016 Will Become Mainstream
2016 was a big year for ISO 13485. A revision to this standard was long overdue and published in March 2016.
There is a three-year grace period to transition from ISO 13485:2003 to ISO 13485:2016. But you should expect ISO 13485:2016 to begin to take hold in 2017.
Prediction: ISO registrars will begin transitioning certifications to ISO 13485:2016 in 2017.
In fact, I know of several ISO audits being scheduled for companies in early 2017 where the registrar is requesting that the 2016 “upgrade” take place.
Beware. Do not just assume that your QMS is already in alignment with ISO 13485:2016. This new revision to the standard does include several changes.
I recommend that you conduct a gap analysis of your current QMS against ISO 13485:2016 and define a quality plan for making this transition.
4. EU Medical Device Regulations and IVD Regulations Finalized
The European Commission has long been discussing changes that impact the medical device industry.
Currently in EU, medical devices and in vitro diagnostics (IVDs) are governed according to directives (MDD and IVDD).
Prediction: EU will finalize and implement medical device regulations (MDRs) and IVD regulations (IVDRs) in the first quarter of 2017.
Yes, these new EU regulations will have an impact on medical devices and IVDs sold in the European market. Yes, there is a transition period expected for both new regulations. MDRs go into full effect within 3 years and IVDRs within 5 years of adoption.
While this seems like you have plenty of time to adjust, I encourage you to stay on top of these changes. These changes will not only impact new products but also any existing products which are already CE marked.
To get the full picture of EU changes, be sure to check out the on-demand webinars Greenlight Guru has on these topics:
- How to Prepare for the New EU Medical Device Regulations (MDR)
- How to Prepare for the New EU In Vitro Diagnostics Regulations (IVDR)
5. Expect FDA 510(k) Review Times to Increase
In the U.S., the most common pathway to receive FDA market clearance for a medical device is a 510(k) submission.
The 510(k) process is well-established and defined. In recent years, the FDA instituted new policy for 510(k)s known as the “refuse to accept” policy.
As a result of implementing this policy, 69% of 510(k)s were rejected during the first half of 2015.
Prediction: FDA 510(k) review times will continue to increase in 2017.
While the final 2016 data is still not available, I would like to share some trends from 2015.
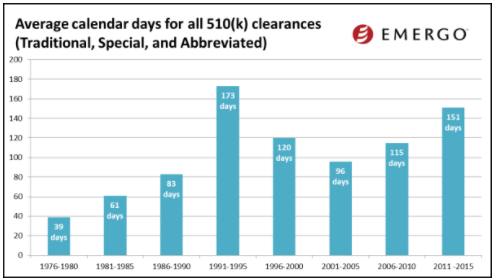
(image courtesy of Emergo Group)
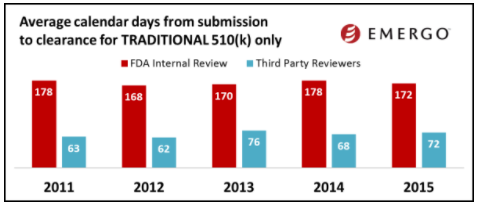
(image courtesy of Emergo Group)
The charts above show how 510(k) review times have been trending over the past several years. And by the looks of this data, you might conclude that 510(k) review times have been fairly consistent over the past few years.
But keep in mind this data does not factor in those 510(k)s that are rejected. When this happens, the clock starts over.
Those are my 5 predictions I believe the medical device industry should expect to happen in 2017.



