
When EU MDR entered into force on May 26, 2017, it raised the bar for what constitutes sufficient clinical evidence to demonstrate a device’s conformity with EU regulations.
Fortunately, the European Commission (EC) provided a transitional period, giving manufacturers with medical devices already on the market time to prepare for MDR requirements.
And to clarify the clinical evidence manufacturers of those legacy medical devices would need to demonstrate conformity, the EC released MDCG 2020-6 - Regulation (EU) 2017/745: Clinical evidence needed for medical devices previously CE marked under Directives 93/42/EEC or 90/385/EEC.
This article will explain what MDCG 2020-6 entails and the types of medical devices to which it applies.
What is the scope of MDCG 2020-6?
MDCG 2020-6 applies to legacy medical devices, meaning those which were CE marked under the European Medical Devices Directive 93/42/EEC (MDD) or Active Implantable Medical Devices Directive 90/385/EEC (AIMDD).
Under current EU MDR timeline set out by the EC, all devices placed on the market before May 25, 2017 were required to be certified under MDR by May 27, 2022. However, devices that received their CE marking under the MDD or AIMDD after May 25, 2017 will need to be recertified within five years from the initial date of certification or by May 27, 2024—whichever comes first. This graphic lays it out well:
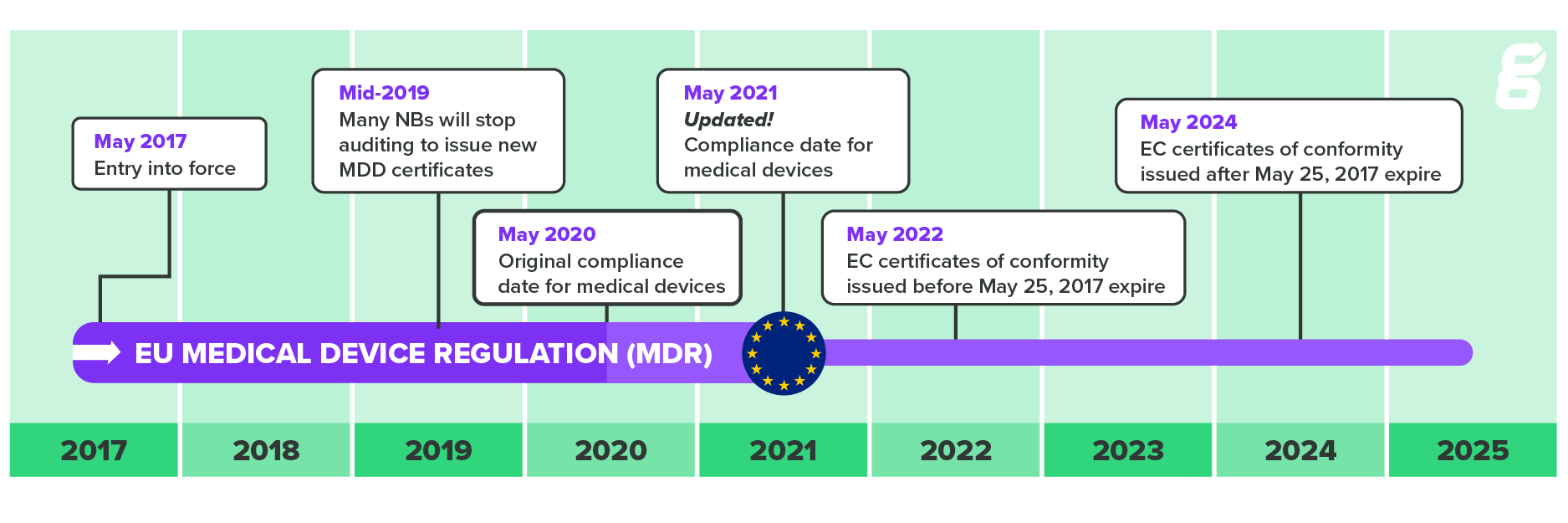
So, there are still devices on the market which have not yet been certified under MDR, but will need to be by 2024 at the latest. To remain on the EU market these devices will need to demonstrate conformity with all relevant General Safety and Performance Requirements (GSPRs) outlined in MDR.
MDCG 2020-6 provides guidance for the manufacturers of these devices on the clinical data they will need to prove that their devices conform with those GSPRs.
What constitutes sufficient clinical evidence?
The term “sufficient clinical evidence” is used in MDR, yet nowhere in the regulation do the authors define the term.
MDCG 2020-6 attempts to fix this oversight in section 5, General Aspects. The guidance states,
Therefore, “sufficient clinical evidence” is understood as “the present result of the qualified assessment which has reached the conclusion that the device is safe and achieves the intended benefit.
At first glance, this definition seems… less than helpful. It somehow doesn’t offer any specifics on what constitutes sufficient clinical evidence for a given device. Instead, it places the emphasis on the results of the “qualified assessment”, meaning the clinical evaluation.
But in section 6.1, the guidance clearly states that part of the clinical evaluation plan is determining the level of clinical evidence your device will require:
The level of clinical evidence required for the device under evaluation needs to be determined by the manufacturer and verified by the notified body. The level of clinical evidence shall be appropriate in view of the characteristics of the device and its intended purpose.
When you consider the vast number of medical devices and their various intended purposes and risk levels, the murky definition of sufficient clinical evidence begins to make sense. It has to be determined by the manufacturer for each device and then demonstrated through the clinical evaluation.
What does MDCG 2020-6 say about clinical evaluation?
MDCG 2020-6 doesn’t offer guidance on the process or methodology for carrying out a clinical evaluation.
However, it does include two appendices that will be helpful in planning and carrying out a clinical evaluation for a legacy medical device:
-
Appendix 1 outlines which sections of MEDDEV 2.7/1 Rev. 4—which offers guidance on the general process for conducting a clinical evaluation—are still relevant for legacy devices under MDR.
-
Appendix 2 lays out the minimum elements that a modified clinical evaluation plan for legacy devices should include.
It also includes a lengthy section (section 6) on the specific aspects of clinical evaluation pertaining to legacy devices, including establishing or updating your clinical evaluation plan.
However, the bulk of section 6 is given over to clinical data. MDCG 2020-6 states that the basis for the clinical evaluation will be post-market clinical data and clinical data generated for the conformity assessment under MDD/AIMDD. Basically, you’ll need the clinical data used to get the device on the market and the clinical data that has been gathered since then.
Accordingly, it offers guidance on identifying, appraising, and analyzing clinical data, as well as generating new clinical data for your legacy medical device, if necessary.
Note that the third appendix in MDCG 2020-6 provides a suggested hierarchy of clinical evidence for confirmation of conformity with GSPRs. This should be especially helpful as you begin gathering and appraising clinical data.
MDCG 2020-6 is essential reading for any medical device manufacturer with legacy medical devices on the EU market, especially if you have questions regarding the clinical evidence you’ll need to demonstrate conformity with the regulations.
Manage all your clinical data and clinical evaluation activities on one purpose-built platform
Clinical evaluation under MDR is not a one-time occurrence. The regulations make it clear that this is an ongoing process that has to occur regularly throughout the lifecycle of your device.
And while that may seem daunting, there are ways to streamline the process and ensure that you have a single source of truth for all your data and documentation. Greenlight Guru’s MedTech Lifecycle Excellence Platform is purpose-built for medical device companies to streamline and manage all quality activities.
If you’re ready to ditch the spreadsheets and generic quality system solutions, then get your free demo of Greenlight Guru today.
BONUS RESOURCE:
Clinical Evaluation Procedure Template
Clinical Evaluation Procedure Template






