
How confident are you when it comes to design validation? Does this always involve clinical evaluation, or not?
We’ve found that, like many other terms in medical device development, the two can end up getting confused in the minds of many companies. When do you use one or the other?
There tends to be a lack of clarity out there because you’ll get different answers, depending on who you ask. Sometimes it all comes down to context, which we’ll take a look at.
Let’s take a look at the two:
General Definitions
When we’re asked about design validation and clinical evaluation, here’s how we look at them in general terms:
Design validation - This is a key requirement for proving that your medical device meets the needs of the defined end-user. It is an element of design controls and is really about proving that the product developed is the correct device in the first place.
When it comes to design validation, it really is a reflection of the work you should have done very early on. For example, you should have gathered user stories and done a thorough job of identifying user needs. Essentially, you’re answering the question, “did we build the right device?”
Clinical evaluation - This has a strong relationship with design validation. In fact, clinical evaluation might be used as a means of validating the device. The twist here is that clinical evaluation can have different meanings depending on who you ask. For example, under the same umbrella, you might find clinical investigation, testing or usage.
In some cases, the clinical evaluation might have nothing to do with your specific product. An evaluation might have been conducted on other devices that are predicate or similar technologies. These would have similar uses to your device so that you can use the evaluation to prove that yours will meet standards.
Clinical evaluation is applicable to your particular product. You may perform actual or simulated use testing. It is multi-layered - you need to understand the intent that the evaluation is addressing. Is it actual or simulated use of your device with test subjects, clinicians or end users? What is included?
The key differentiator between the two terms is that design validation is part of design controls and is a required part of the product development process prior to transferring the product into full production. Clinical evaluation is used to support design validation activities, but it won’t always be required.
Clinical Evaluation for a Technical File
If you’re wanting to market your medical device in Europe, then you will be required to write and maintain a Clinical Evaluation Report (CER). This is part of your technical file submission. The EU has very specific requirements for clinical evaluation documentation and reviews. An evaluation can include devices that have similar intended uses to your device and meet relevant essential requirements.
This is all part of being issued a CE Marking Certificate for the EU, but you are still required to maintain that CER once you have the certificate.
What is CE Marking?
You’ve probably seen this marketing before although you may not have realized it, the CE Mark is a symbol stamped onto approved products and is an abbreviation of the French phrase "Conformité Européene", (European Conformity).
When a manufacturer stamps their product with this mark, they are declaring that the product is compliant with essential European regulations. The mark also:
- Indicates to government officials that the product may legally be placed on the market in their country
- Allows for the free movement of the product within the EU
While it is the manufacturer’s responsibility to affix a CE marking to their device, it is also their responsibility to ensure they are compliant with the regulations for keeping it. You can’t just slap one on your medical device without having gone through the steps to compliance, otherwise penalties will apply.
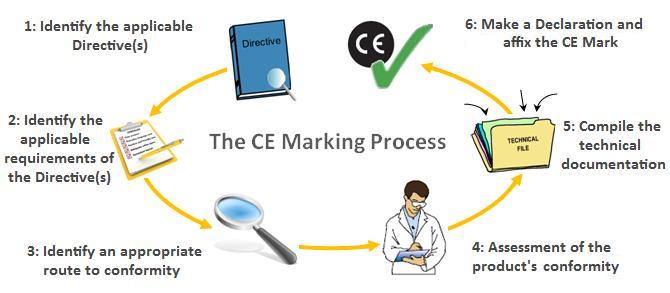 The CE Marking Process, via CE Marking Association
The CE Marking Process, via CE Marking Association The Technical File
As you can see at step 5 in the above diagram, compiling your technical documentation (or technical file) is an essential part of the CE marking process. For medical devices, the technical file must consist of the following to satisfy the criteria of the Harmonized Standards:
- Product description and specifications
- Manufacturing information
- Risk management file
- Design verification and validation test reports
- Clinical evaluation
- Labeling
As you can see from this, your clinical evaluation is just one part of the requirements for your technical file.
What does the CER involve?
The clinical evaluation report documents the entire process of your clinical evaluation. This evaluation involves the assessment and analysis of clinical data for your device to verify its safety and performance. The data used may be both pre and post-market that is relevant to verify intended use. This includes data specific to the device as well as any data relating to devices claimed as equivalent by the manufacturer.
Cromsource provides a table below which outlines the clinical data sources for a clinical evaluation:
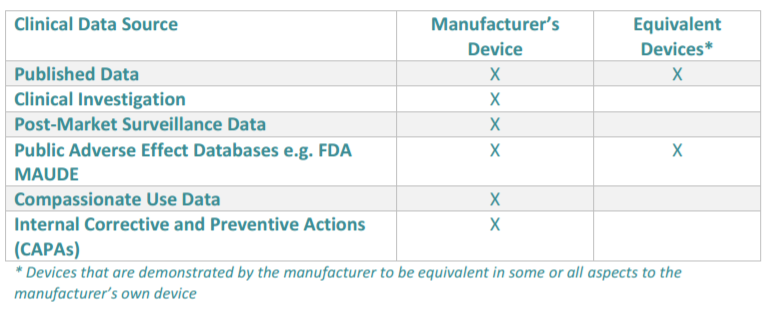
Here is how the evaluation works, as taken from Cromsource:
The clinical evaluation needs to cover: any design features that pose special performance or safety concerns; the intended purpose and application of the device; and the specific claims made about the clinical performance and safety of the device. It is important to describe the merit and limitations of any data cited or included in the evaluation.
The manufacturer’s risk assessment documentation is included in the review process to ensure that all risks identified are discussed and addressed/mitigated in it. The instructions for use (IFU) for the device are reviewed during the process to ensure that data is gathered from the same population using the device in the same way for the same indications, as described in the IFU. Finally, conclusions are drawn about whether the Essential Requirements relevant to clinical safety and performance are met.
Recent EU changes
You may have heard other manufacturers commenting that the EU CE Mark process is simpler and more predictable than the FDA; as Thomas Jull discusses, the tune is set to change with some recent updates made to EU regulations.There’s a shift more toward the FDA way of doing things and those changes will affect new devices and those that already bear the CE Mark. There are new rules on classification and heavier post-market surveillance requirements, among others, meaning more emphasis on post-market clinical evaluation.
“Post-Market Clinical Follow-Up (PMCF) must be planned in, as the information gathered will be used to analyze the safety and performance of the device.
This, for most device classes, will now be reported annually to the authorities. (See Annex XIII, Part B, 2.2, of the MDR for what needs to be included in the plan.) Along with the aims of this system to reduce risks, unannounced audits are expected to become more common and frequent.”
Browse through Jull’s article for Medical Design Technology Magazine for more information on the changes.
An ongoing process
Getting back to design validation and the inter-relationships with clinical evaluation, as we determined earlier, you might use clinical evaluation as part of your design validation, which is an activity that happens pre-market.
However, overall, clinical evaluation is something that is an ongoing process throughout the life cycle of a medical device. Once you’ve completed the initial evaluations that help get your CE Mark or help you to meet the requirements of other regulatory authorities, there will be occasions to reevaluate the clinical evaluation as new clinical information becomes available (such as from studies or use) or as any changes are made to the device.
As we’ve seen with the updating of regulations, there is an expectation that you will continue to gather clinical data and use it to prove the safety and effectiveness of your product.
The Bottom Line: Safety and Effectiveness
The differences between design validation and clinical evaluation lie in the context under which the evaluation is conducted. While design validation is a pre-market requirement to demonstrate that you’ve developed the correct product for users, clinical evaluation might be part of that demonstration.
Clinical evaluation is taken further as it is used under several other circumstances, including for your technical file if you’re seeking a European CE Mark. Unlike design validation, clinical evaluation is a lifecycle activity which continues well into post-market.
The overall aim is, as always, to prove that you have a safe and effective device.
Still using a manual or paper-based approach to manage your design controls or quality processes? Click here to learn more about how Greenlight Guru's modern eQMS software platform exclusively for medical device companies is helping device makers all over the globe in more than 320 cities and 26 countries get safer products to market faster with less risk while ensuring regulatory compliance.





