See Your ROI with Greenlight Guru in Seconds


Experience the #1 QMS software for medical device companies first-hand. Click through an interactive demo.

The pursuit of MedTech Lifecycle Excellence requires that you consider the entire lifecycle of your medical device. This includes systematic monitoring of your device after it’s been placed on the market—in other words, postmarket surveillance (PMS).
Postmarket surveillance is vital to demonstrating that your device continues to be safe and effective after it’s gained regulatory approval. This is especially important for implantable devices.
Whether you’re wondering how to get started with PMS for your medical device, or just have a specific question you need answered, this guide is for you. We’re going to cover the basics of postmarket surveillance, as well as the regulations that govern it in both the US and the EU. Finally, we’ll wrap up with some best practices and actionable tips for carrying out your own postmarket surveillance activities.
We’ve got a lot to cover, so let’s get started.
|
Why is postmarket surveillance so important? |
|
Postmarket surveillance activities under 21 CFR Part 822 |
|
What are the regulations governing postmarket surveillance in the EU? Clinical evaluation report (CER) |
|
What other guidance documents, standards, and resources can help with postmarket surveillance? MDCG guidance documents covering postmarket surveillance |
|
Best practices for carrying out postmarket surveillance Start working on postmarket surveillance early |
Postmarket surveillance is the means by which medical device manufacturers monitor their devices while on the market. It’s the systematic generation and collection of information on the device and its real-world use, which manufacturers can then utilize in a number of ways, including:
Discovering safety issues with the design or use of the device
Accurately understanding how the device is used once it’s on the market
Gathering clinical evidence on device use in the market, to promote commercial use cases or improve product and services
Gathering data for the production of iterations or new devices
Complying with regulatory requirements
In the strictest sense, postmarket surveillance is necessary because it’s required by regulators around the world. You are required to follow your product throughout its entire lifecycle, which includes the device’s time on the market.
But bare bones compliance with regulations shouldn’t be the only reason you take PMS seriously. The reason regulatory bodies, like FDA, place such an emphasis on PMS is because problems with a device can affect patients’ safety.
If you aren’t taking your postmarket surveillance duties seriously enough, you could miss indications of a safety issue with your device that lead to a catastrophic outcome, like a patient’s death, or other serious consequences, like a delayed surgery.
Even low-risk failures have consequences. Your company’s reputation is on the line every time someone uses your products. It doesn’t take many failures to teach a user they can’t rely on your product—and by extension, your company.
Postmarket surveillance isn’t just about avoiding poor outcomes, however. There are a host of benefits to both patients and businesses when manufacturers use PMS proactively.
For instance, the information you gather as part of your PMS activities is invaluable when it comes to developing new products or improving existing systems. You’ll have years of data at your disposal to help you innovate, avoid previous mistakes, simplify manufacturing, and generally make the path to market easier and faster.
Your postmarket surveillance activities can help you answer questions like:
Is our device too difficult to manufacture?
Do we need to change materials?
Do we need to change a supplier who isn’t meeting our standards of quality?
Can we market our device differently, using post-market clinical evidence?
Can we use reimbursement pathways we didn’t before?
For example, let’s say you make a hip implant and rely on several different suppliers for the parts of the implant. After the device is on the market, you receive a complaint from a surgeon that there was white residue on the device before implantation. You investigate the complaint and find that your supplier controls are the root cause of the issue and determine your incoming inspections need to be more critical.
Without a strong postmarket surveillance system in place, that complaint may never get back to you or it may get lost within your organization—meaning you’ll miss the opportunity not only to fix the issue, but to improve your supplier controls and prevent future problems.
Too many medical device companies make postmarket surveillance an afterthought, and as a result, they can’t become proactive about it. They’re stuck firefighting as complaints and nonconformances roll in, and they never see the full benefits of a great PMS system.
Part of the reason this happens is because companies aren’t using the right tools to manage their quality processes. As we’ll see, there is an enormous amount of potential PMS data out there, but it’s only useful if you can easily collect and analyze it.
When companies use paper-based tools like Excel or other generic software, more work goes into managing the tools and staying compliant than translating the data into meaningful information for postmarket surveillance.
Both FDA’s Quality System Regulation (QSR) and ISO 13485:2016 include requirements for post-market processes in your company’s quality management system (QMS).
The QMS processes that touch on postmarket surveillance include:
Complaint handling
Nonconformance management
Corrective Action and Preventive Action (CAPA)
Internal auditing
If used correctly these processes and the data they generate should provide you with a strong foundation for postmarket surveillance. And this is where an interconnected and intuitive eQMS platform comes into play.
The data from your post-market processes is only valuable when it’s being used. To get the most out of it, you need to analyze the data that comes in, studying trends and identifying whether you have repeated issues that need closer examination. This ties directly into your risk management and answers question like:
Does the actual rate of adverse events track with the anticipated rate?
What about the rate of nonconformances?
How large are the discrepancies and what is the cause of them?
Are you seeing adverse events that you didn’t have in your risk documentation?
Not only may your risk documentation need to be updated in light of the real-world evidence of your product, but you may need to take further action like opening a CAPA or even initiating a field safety corrective action (FSCA), like a recall. The interplay between areas like risk, quality, and design and development highlights just how important it is for your team to be working in a single, connected system.
In years past, one of the problems that plagued medical device companies was the lack of purpose-built tools for managing quality processes. If you were bringing a device to market, your QMS either lived in a hodge-podge of disconnected software, a generic eQMS that had to be fully customized and validated, or (most frightening of all) a bunch of filing cabinets sitting in a windowless room somewhere.
Today, you have another option. A better option. Greenlight Guru’s eQMS platform is built specifically to give MedTech companies an end-to-end system to manage the entire product lifecycle all in one connected place. Purpose-built post-market workspaces for CAPAs, nonconformances, complaints, and audits mean all your PMS data is readily available for analysis and action.
To see how a purpose-built QMS can streamline your processes and help maintain traceability, get your free demo of Greenlight Guru’s eQMS today.
Having strong post-market quality processes in place is essential, but the QMS requirements are not the end of postmarket surveillance in the US or the EU.
The regulatory bodies in the US and the EU take different approaches to postmarket surveillance of medical devices. In the US, your postmarket surveillance is mostly encompassed by the quality processes in your QMS that we just went over—nonconformance, complaints, CAPA, and internal auditing.
There are, however, other potential postmarket surveillance activities that FDA may require from your company beyond your quality processes. These can be found in 21 CFR Part 822 - Postmarket Surveillance. The regulation provides:
procedures and requirements for postmarket surveillance of class II and class III devices that meet any of the following criteria:
If your device falls into any of these categories, FDA has the power to order your company to conduct postmarket surveillance of your medical device(s) using a formalized plan under section 522 of the Federal Food, Drug, and Cosmetic Act.
Keep in mind, if FDA orders you to conduct postmarket surveillance, these PMS activities are in addition to the quality processes we’ve discussed, such as nonconformance and CAPA. You are not absolved from carrying out these quality processes just because FDA has not issued you a letter under section 522.
Part 822 of the regulation also states:
We will notify you as soon as we have determined that postmarket surveillance of your device is necessary, based on the identification of a surveillance question. This may occur during the review of a marketing application for your device, as your device goes to market, or after your device has been marketed for a period of time.
In a nutshell, FDA can order you to perform postmarket surveillance activities using a formalized plan at any time. If ordered, you will need to send them a plan for collecting data that will answer the questions they want addressed within 30 days of the date you receive their order. You also have 15 months from receiving the FDA’s letter to begin the surveillance activity.
FDA has put out a guidance document on the subject: Postmarket Surveillance Under Section 522 of the Federal Food, Drug, and Cosmetic Act - Guidance for Industry and Food and Drug Administration Staff.
The guidance document lists a number of different types of postmarket surveillance designs you may need to use to address the surveillance question(s) the agency has raised. These include:
Randomized clinical trial
Prospective cohort study
Retrospective cohort study
Cross-sectional study
Enhanced surveillance
Active surveillance
Meta-analysis
Prospective & retrospective study
Case control study
Non-clinical study
Animal study
Other designs that don’t fall into one of these categories
The likelihood that you will receive a letter from FDA to perform specific postmarket surveillance activities is different for every company and device. Generally speaking, there will be an event that triggers a PMS order from FDA, such as a recall. But they may also ask for certain PMS activities after your 510(k) or PMA submission if they identify a surveillance question(s) that needs to be answered.
FDA may also require what it calls “post-approval studies” if your device is approved via a Premarket Approval Application (PMA) or Humanitarian Device Exemption (HDE). In these cases, when FDA approves your device, they may also inform you that you’ll be required to carry out a study (or studies) to help ensure the safety and effectiveness of the device while it’s on the market.
Because your quality processes play such a substantial role in postmarket surveillance in the US, the best advice we can offer is to get them in place early.
Don’t wait until your device has been approved to start thinking about your post-market responsibilities. You should be working on these processes during design and development, so that when your device does hit the market, you’re prepared to gather post-market data and act on it in a timely fashion.
However, if you are ordered to perform a specific PMS activity by FDA, then you’ll need more than just your QMS to capture the data from those studies. You’ll need a validated, compliant, and MedTech-specific electronic data capture (EDC) system—like the one we offer at Greenlight Guru.
Get your free demo of our EDC platform and see how a modern, purpose-built platform can streamline your clinical data collection and management.
Postmarket surveillance requirements in the EU are significantly different from those in the US. The EU has a more structured and explicit postmarket surveillance program that is outlined in Chapter VII of EU MDR.
You must have the same quality system processes in place that we’ve already discussed (as required by ISO 13485:2016). But whereas those quality processes encompass most of your postmarket surveillance activities in the US, they’re just the beginning of what will be required of you in the EU.
Under EU MDR, there have been significant additions to the postmarket surveillance and vigilance activities required of medical device manufacturers. Manufacturers with devices that were CE marked under the old Medical Device Directive have been wrestling with the new PMS requirements for years now.
But if you’re planning on entering the EU market, then it’s a good idea to start learning about the postmarket surveillance requirements and planning for them ASAP.
These PMS requirements can be found in Chapter VII of both EU MDR, and in this section, we’ll outline the most important documentation requirements. Your device class will dictate the documentation you are responsible for and how often you’ll need to update those documents.
The following table will help clarify the responsibilities based on device class under EU MDR.
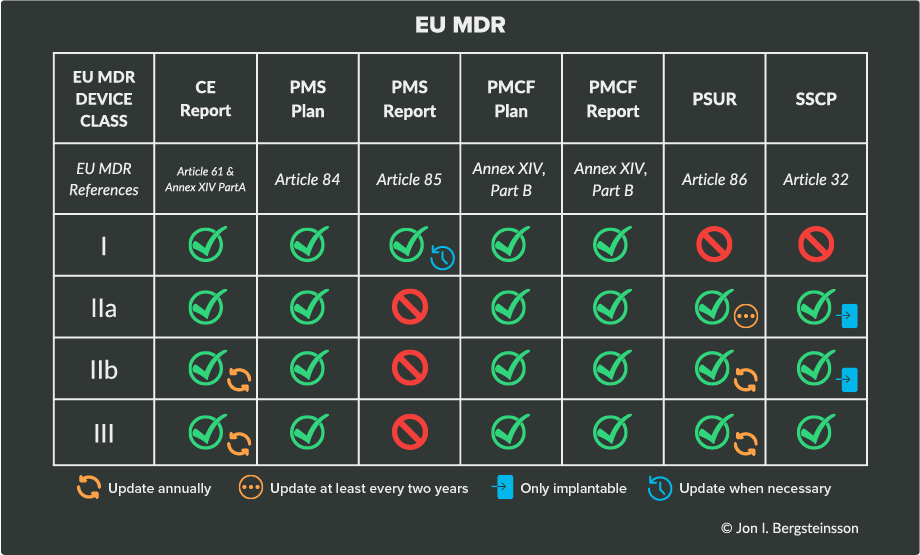
Here’s a breakdown of each of the items that EU MDR requires:
What is it? The clinical evaluation report (CER) is documentation of the clinical evaluation required of every medical device sold in the EU. Its purpose is to prove your device performs as intended without compromising the safety of its end users.
The CER is meant to be a living document, and if at any point you receive new and pertinent information about your device through postmarket surveillance or new clinical evaluations, then the CER must be updated to reflect that. However, even if you don’t receive new information, your clinical evaluation report must still be updated according to a schedule that you, the manufacturer, define and justify.
What devices require it? The CER is required for every class of medical device. For devices without significant risk, the CER must be updated every two to five years. The schedule you choose should reflect the risk classification of your device and how well-established the technology behind the device is.
What is it? The PMS plan is part of your device’s required technical documentation and details your strategy for continuously monitoring and collecting data and safety information on the device.
The plan is part of the requirements for a PMS system, and is intended to outline the criteria for the benefit-risk assessment of the device and processes for:
Collecting and analyzing data
Addressing submitted complaints
Communicating data to regulatory bodies and users
Implementing corrective actions on devices
Additionally, the PMS plan is used to determine whether or not a post-market clinical followup (PMCF) is required.
Which devices require it? The PMS plan is another document that is required of every device class. The PMS plan must be established and documented prior to placing the medical device on the EU market for the first time, and updated as necessary during its lifecycle.
What is it? A postmarket surveillance report (PMSR) is a summary of the results and conclusions drawn from the data generated as part of the PMS plan. As part of a device’s technical documentation, it primarily serves as proof of your compliance with MDR’s requirements for postmarket surveillance.
Article 85 of EU MDR states that a PMSR must contain information summarizing the results and conclusions of the post-market data generated by the PMS plan. It also must be presented with a rationale and description for any preventive and corrective actions taken.
Which devices require it? Postmarket surveillance reports are used only for Class I devices, as opposed to the periodic safety update report required for higher-risk device classes
The PMSR can be updated whenever the manufacturer considers it necessary and/or delivered upon request by an authority. But that means it must be produced/updated from time to time, and it’s recommended to do so at least once every three years
What is it? The periodic safety update report is a summary of the results of postmarket surveillance activities as well as the conclusions that manufacturers have drawn from those results. If the manufacturer has taken any corrective or preventive actions (CAPAs), a description and rationale for the actions must also be included in this report.
PSURs are identical to PMSRs with a few additions—manufacturers must publish the conclusion of the benefit-risk determination, main findings of the post-market clinical or performance follow-up, sales volume, and estimated user population characteristics and usage frequency.
Which devices require it? The PSUR is required for Class IIa, IIb, and III medical devices. The periodic safety update report is part of a device’s technical documentation, and it has to be updated throughout the device’s lifecycle. The PSUR is initially submitted to a Notified Body during the device’s conformity assessment audit, but from then on it must be updated either annually or biennially.
What is it? The PMCF requires manufacturers to proactively collect and evaluate clinical data on a device’s performance and safety.
The goal of PMCF is to confirm existing clinical claims and identify possibly unknown side-effects, assess emergent risks, and prevent off-label misuse. This data can be captured through general PMCF activities, such as gathering feedback from end users and information from scientific literature.
However, in order to substantiate the data, it often is necessary to use specific PMCF activities—higher-level operations which can be used to scientifically illustrate safety and clinical performance, often based on case-specific data.
These post-market clinical followup activities include:
PMCF studies (or observational or non-interventional clinical investigations)
Post-market interventional clinical investigations
Evaluation of data from suitable registries
Investigator initiated studies
Case cohorts and case series
Other subject-specific clinical data collection activities
To satisfy the PMCF requirements, you will need both a PMCF plan and a PMCF report. You can learn more about structuring your plan and report in our Guide to PMCF Activities.
What devices require it? The PMCF is required for all device classes in the EU. It is possible that your device may not require a PMCF, but you will be required to justify your decision not to carry one out.
What is it? The summary of safety and clinical performance is an external document that incorporates information related to a medical device, such as general information, a summary of the clinical data collected from the device, or possible therapeutic alternatives.
The main purpose of the SSCP is to provide both healthcare workers and patients access to current clinical, safety, performance, and other types of data from the medical device, as this aligns with the MDR philosophy of providing a more transparent and robust regulatory framework.
What devices require it? The SSCP is intended for devices which are higher risk or invasive, specifically:
Implantable Class IIa devices
Implantable Class IIb devices
All Class III devices
The European Union Database on Medical Device (EUDAMED) is a digital exchange for legal information on medical devices under EU MDR.
The goal of EUDAMED is to improve transparency and help coordinate information related to medical devices and diagnostics that are on the EU market. The database is made up of six interconnected modules:
Actors registration
UDI/Devices registration
Clinical investigations and performance studies
Notified bodies and certificates
Vigilance and postmarket surveillance
Market surveillance
At the time of writing, only the modules on actor registration, UDI/device registration, and notified bodies are available for voluntary use. The modules on clinical investigations and performance studies, vigilance and postmarket surveillance, and market surveillance will be released once the entire system is functional, which is currently scheduled for the end of 2024.
At that point, use of the system will become mandatory, so it’s a good idea to become familiar with the system and now and even begin using the three modules that are available for voluntary use.
The requirements for postmarket surveillance under MDR and IVDR can seem daunting, especially if your device requires many of these plans, reports, and post-market clinical activities to demonstrate sufficient clinical evidence of its safety and effectiveness.
Two big pieces of advice here: start early and use the right tools.
EU MDR has made it much more difficult to claim equivalency with other devices in the EU, which means more manufacturers may need to generate their own clinical data for the PMCF.
As you look for the best way to manage your clinical data, you’ll notice that most of the electronic data capture (EDC) tools you come across are primarily older, pharma-specific tools that require intense customization.
At Greenlight Guru, our goal is to provide MedTech companies with tools built specifically for their needs. That’s why our modern, validated EDC platform is designed specifically for medical device manufacturers—letting you quickly build studies, adjust designs, and be confident in your data collection’s compliance with EU and US regulations.
Get your free demo of our EDC platform and see how a modern, purpose-built platform can streamline your post-market clinical data collection.
Clearly, there’s a lot for manufacturers to learn and begin thinking about when it comes to postmarket surveillance. It can be an overwhelming topic, especially when you begin looking at the many EU regulatory requirements.
Fortunately, the regulations are not the only resources out there to help you understand how to set up a successful postmarket surveillance program. There are guidance documents and resources put out by regulatory bodies and standards from independent organizations like ISO, that can be enormously helpful.
The Medical Device Coordination Group has put out over 100 guidance documents on EU MDR implementation, serving as indispensable resources for manufacturers who need to comply with postmarket surveillance requirements.
Some of the most applicable MDCG guidances to postmarket surveillance include:
MDCG 2020-7 contains a template and guidance for your PMCF plan.
MDCG 2020-8 contains a template and guidance for your PMCF evaluation report.
MDCG 2019-9 Rev. 1 contains guidance on each of the required sections of the SSCP document required for high risk devices in the EU.
ISO 20416:2020 is an international standard for postmarket surveillance. The document provides a proactive and systematic process for manufacturers to collect and analyze post-market data.
ISO states that the process outlined in ISO 20416:2020 is consistent with both ISO 13485 and ISO 14971. So, if you’re following these standards (which you should be!) you should be able to integrate the postmarket surveillance process outlined in ISO 20416 into your quality and risk management processes.
Keep in mind that while this standard is meant to help you meet regulatory requirements, it does not replace those requirements, and implementing the process in the standard does not exempt you from other post-market requirements, such as those mandated in the EU.
Team Notified Body is an association of notified bodies in Europe working in the medical device space. The association regularly puts out position papers that offer a unified approach for notified bodies on various subjects related to their work.
This helps ensure that the best practices of various notified bodies, which are independent organizations, don’t vary too widely. From a manufacturer’s perspective, these position papers offer a great insight into what notified bodies will expect.
In the Team Notified Body Position Paper Best Practice Guidance for the Submission of Technical Documentation under Annex II of the Medical Device Regulation (EU) 2017/745 you’ll find information on what post-market data and information a notified body will be looking for.
Don’t be put off by lengthy or odd titles; there’s excellent information in these documents!
The Manufacturer and User Facility Experience (MAUDE) database is where FDA collects medical device reports of adverse events related to medical devices. The database houses records of “suspected device-associated deaths, serious injuries and malfunctions” that are reported by mandatory reporters such as manufacturers, importers, and device facilities as well as voluntary reporters like health care professionals and patients.
Not every adverse event may make it back to you as a complaint; instead they may be reported and logged in the MAUDE database. If your device is implicated in a medical device report in the MAUDE database, you will receive notice from FDA, but it may not be in the most timely manner.
So, it’s a good idea to keep an eye on the MAUDE database, not only for adverse events related to your own devices, but also those of similar devices or competitor’s devices that have the same indications for use.
Many medical devices being brought to market today use software that can pose cybersecurity risks. If your device uses software or is software as a medical device (SaMD), it is incumbent upon you as the manufacturer to monitor, identify, and address cybersecurity risks as part of your post-market management of your medical device.
Addressing cybersecurity will interact with the post-market quality processes we’ve been discussing. FDA’s guidance document states:
It is essential that manufacturers implement comprehensive cybersecurity risk management programs and documentation consistent with the Quality System Regulation (21 CFR part 820), including but not limited to complaint handling (21 CFR 820.198), quality audit (21 CFR 820.22), corrective and preventive action (21 CFR 820.100), software validation and risk analysis (21 CFR 820.30(g)) and servicing (21 CFR 820.200).
The guidance document also includes an appendix that outlines the elements of an effective postmarket cybersecurity program.
While a comprehensive discussion of this topic is beyond the scope of this guide, you can find more information in our Ultimate Guide to Software as a Medical Device, as well as these episodes of the Global Medical Device Podcast:
Episode 264: Approaching Cybersecurity & Usability as a SaMD Company
Episode 270: Shifting Sands of SaMD Cybersecurity Regulations
And if you’re looking to learn more independently, Greenlight Guru Academy offers courses on cybersecurity, SaMD, and IEC 62304 - Software lifecycle processes for medical devices.
Every manufacturer’s postmarket surveillance activities will look a little different. However, there are some common challenges and mistakes that companies run into over and over.
Whether you’re looking to build out your postmarket surveillance program, or want to make changes to improve what you already have, following these best practices will save you time and money and ensure patient safety. This will allow you to keep your device on the market for the long haul.
We can’t stress enough how important it is to start thinking about postmarket surveillance as early as possible. The previous sections of this guide should give you an indication of how difficult it will be to play catch up with your PMS program.
Too many companies hear “postmarket surveillance” and think, “We don’t need to worry about that yet; we’re not on the market!” That’s a dangerous stance to take. It’s dangerous for your patients who rely on your devices, but it’s also dangerous to your business.
Look at this list of the top reasons for FDA findings during inspections from 2017 to 2021. Problems with the CAPA procedures are the number one reason for citation, followed by complaints and nonconformances. These are fundamental postmarket surveillance processes, but they consistently cause problems for companies that don’t invest in properly managing them.
Failure to take postmarket surveillance seriously from day one is an existential threat to your business, and you should be working on your PMS program early in development—not once the device is ready to hit the market.
The mere existence of a complaint is no guarantee that something will be done about it. From the moment that a complaint is logged—by customer service or a sales rep, for instance—there are many places where communication can break down and that information can get lost or siloed.
For example, if your sales reps aren’t submitting complaints, then you have a problem at the source. But it’s also possible that the complaints are being submitted, but an internal breakdown means they aren’t making it back to the product development team.
At that point, yes, the data has been collected, but it’s not being acted upon. Your product development team may not know they need to make a change to the device’s design or the design of a new device they’re working on, based on a series of complaints.
And a lack of open and timely communication can have a snowball effect. If sales reps are regularly fielding complaints and submitting them, and nothing comes of it, they’re likely to stop sending complaints, making your problem even worse.
Part of the solution can be training—and not just for those who deal with complaints regularly. Everyone in the company should be trained on what constitutes a complaint (yes, even something heard in casual conversation at an event) and how to submit them according to company procedures.
But maybe the best way to ensure your internal processes are being followed is our next tip.
In our 2023 Medtech Industry Benchmark Report, 40% of respondents with products on the market said they were using general purpose tools like paper, Excel, or Word for their quality management system. Another 37% said they were using those general purpose tools for complaint handling, and 44% said they used general purpose tools for clinical testing.
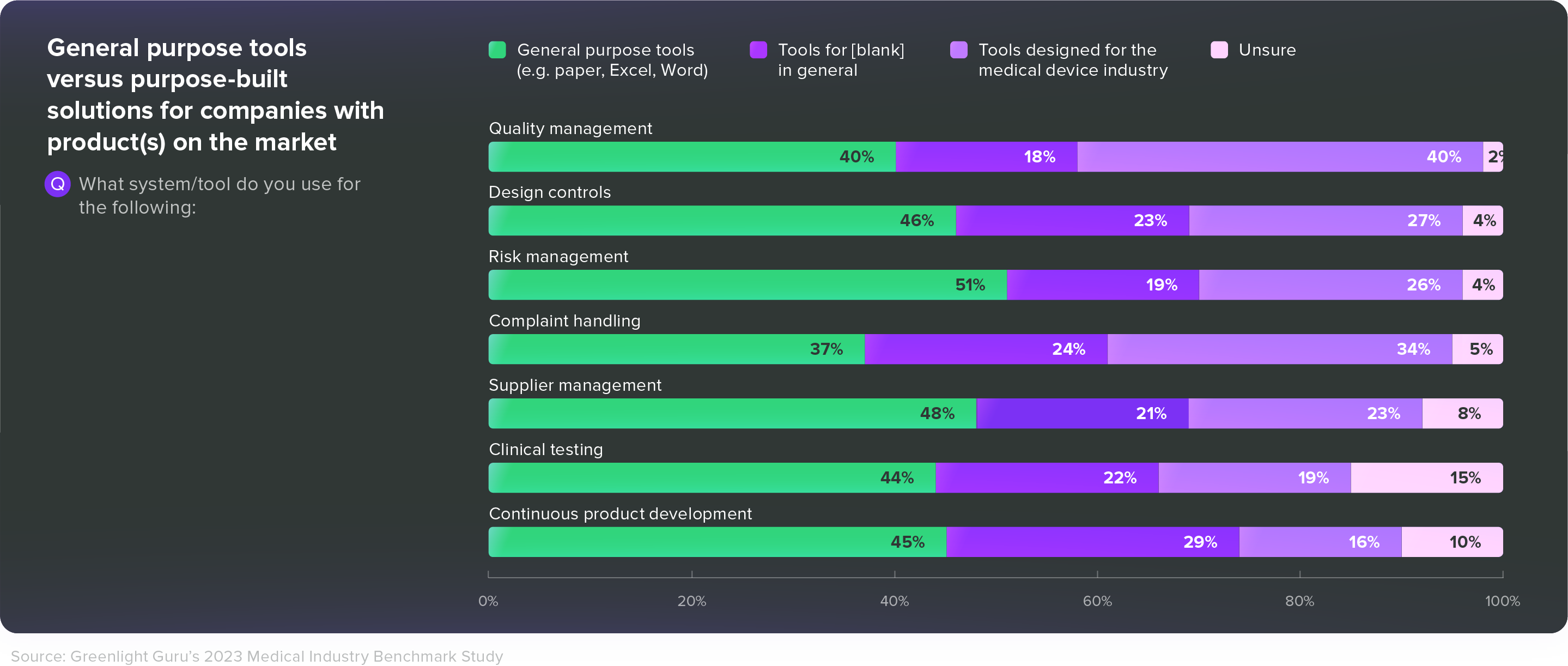
Given all the post-market responsibilities MedTech companies face—and the challenges we’ve just discussed—those are alarming numbers. Medical device companies need tools that are built specifically for them—tools that connect disparate groups across departments and increase efficiency without dictating or disrupting workflows.
At Greenlight Guru, we built our eQMS and EDC platforms specifically for MedTech because we understand the challenges that come with trying to make generic software work for a complex and highly regulated industry.
Here’s how we help you build a best-in-class postmarket surveillance system and ensure all your PMS data is being used.
Automated quality processes. Manually doing something over and over again isn’t just dull and a poor use of time—it introduces countless opportunities for error. Greenlight Guru allows you to automate these quality processes with custom workflow templates built into your QMS, erasing countless hours spent on inefficient administrative work.
You’ll have better visibility and more efficient workflows in all your PMS quality processes:
Task assignment and follow-up. Without purpose-built tools, coordinating investigations is difficult and becomes increasingly complex as the scope of the investigation grows. Overdue action items, missing pieces of evidence, and missing signatures are all common problems that keep investigations lingering for far longer than they should.
But when everyone is using the same single source of truth, investigations become more manageable. Our eQMS allows you to easily assign tasks, track progress, and send reminders to keep the process moving toward completion. Spend less time coordinating and more time getting to the bottom of the problem and correcting it.
Task assignment and follow-up. Without purpose-built tools, coordinating investigations is difficult and becomes increasingly complex as the scope of the investigation grows. Overdue action items, missing pieces of evidence, and missing signatures are all common problems that keep investigations lingering for far longer than they should.
But when everyone is using the same single source of truth, investigations become more manageable. Our eQMS allows you to easily assign tasks, track progress, and send reminders to keep the process moving toward completion. Spend less time coordinating and more time getting to the bottom of the problem and correcting it.
Validated and compliant EDC system. The clinical data you generate from post-market clinical studies is too valuable to risk collecting and housing it in general-purpose tools like Excel. Greenlight Guru’s EDC platform comes pre-validated and is built specifically for medical device clinical trials. Flexible, secure, and compliant, our EDC platform makes it simple to collect clinical data and use it to meet regulatory requirements, get insights from your data, and keep patients safe.
PMCF survey. This is often the choice for many medical device companies to comply with PMCF requirements under EU MDR. To successfully conduct a PMCF survey and pass the scrutiny of Notified Bodies, the sponsor must ensure that data collection follows best practices. Our EDC platform facilitates EU MDR, ISO 14155 compliance via our easily customizable eCRF, surveys and cases.
Adverse Events Reporting. The AE and SAE reporting modules in Greenlight Guru’s EDC platform make it easy for clinical teams to document, categorize, and report on AEs, SAEs, and medical device deficiencies. Our AE module offers straightforward GCP-compliant reporting and customizable forms to fit the needs of any study, with flexible access and permissions, real-time event progress tracking, and AE-specific data export.
Medical device companies that have excellent postmarket surveillance programs have buy-in from everyone in the organization on the importance of quality. This means management understands and actively advocates for quality. This means every department views quality as their responsibility (not just the Quality team).
There is a strong “if you see something, say something” ethos in these companies. Complaints and other post-market data doesn’t slip through the cracks because employees understand their responsibilities and why it’s so important they carry out those responsibilities.
Building a culture of quality doesn’t happen overnight. But it does have to happen if you want to put the safest, most effective medical devices on the market and keep them there for the long run.
If you want to do more than just comply with the regulations—if you’re in the pursuit of MedTech Lifecycle Excellence—then get your free demo of Greenlight Guru today and see how our purpose-built solutions can get you there.
 Brittani Smith, Medical Device Guru, Greenlight Guru
Brittani Smith, Medical Device Guru, Greenlight Guru
Brittani Smith is a Medical Device Guru and Biomedical Engineer who has spent her career in the orthopedic industry working in regulatory, post-market surveillance on the quality team, and remediation engineering work. She also has experience in labeling software and transitioning from paper IFUs to electronic based.
Brittani loves helping customers set up and improve their postmarket surveillance and processes. Brittani enjoys digging into the weeds of all things regulations. She intimately understands how important it is to have a quality over compliance mindset in an organization.
Brittani is equally as passionate ensuring improving quality of life is at the forefront of all she does and being able to help her customers do the same while establishing a QMS and bringing a product to market.
 Alex Naber, Medical Device Guru, Greenlight Guru
Alex Naber, Medical Device Guru, Greenlight Guru
Alex Naber is a Medical Device Guru and Biomedical Engineer who has spent his career in various Quality roles within the orthopedic industry. This has included being the quality lead on development projects, complaint investigation and reporting, CAPA management, Quality Systems management, and managing PMS teams. He is also a Certified ISO 13485 Lead Auditor.
Alex believes having a culture of Quality yields both products that are safe and effective and products that people want to use repeatedly. He is a lifelong learner and loves to use his experience and anything he picks up along the way to help customers develop an efficient QMS that brings quality products to market.














 What is postmarket surveillance of medical devices?
What is postmarket surveillance of medical devices? What are the regulations governing postmarket surveillance in the US?
What are the regulations governing postmarket surveillance in the US? What are the regulations governing postmarket surveillance in the EU?
What are the regulations governing postmarket surveillance in the EU? What other guidance documents, standards & resources can help with postmarket surveillance?
What other guidance documents, standards & resources can help with postmarket surveillance?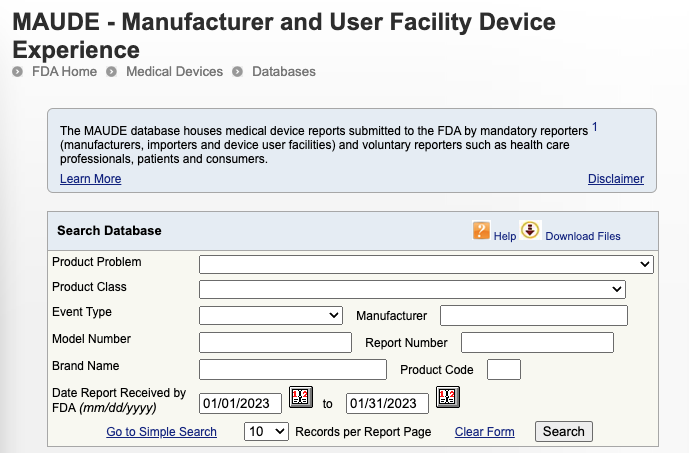
 Best practices for carrying out postmarket surveillance
Best practices for carrying out postmarket surveillance
