See Your ROI with Greenlight Guru in Seconds


Experience the #1 QMS software for medical device companies first-hand. Click through an interactive demo.


What exactly is a risk-based QMS?
This is a timely topic to get into. In 2016, ISO 13485 was updated with one of the key concepts presented being the idea of a risk-based QMS. Historically, regulations have almost exclusively looked at risk in terms of either the design and direct product-related elements, or the manufacturing process.
Design-focused risk was all about what might happen to the patient whereas manufacturing-focused risk is the impact of risk elements on our ability to deliver the product. With design risk, you assume the product was manufactured correctly; with manufacturing risk, you assume it was designed correctly.
The new regulations describe the process behind risk management rather than these more traditional product-focused approaches.
Here are some key areas that medical device developers need in order to apply a risk-based QMS.
You identify all vendors and resources (parts, pieces, services, packaging etc.) that you need to buy in order to manufacture your device. You capture these vendors on an approved supplier list (ASL) that you can share with your team.
Risk has been infused in this process for a long time. Usually you’ll put each supplier in a bucket or category, with the two major categories being “critical” and “noncritical.”
The level of criticality depends on the type of items you have qualified and approved for the supplier and the risk to patients. You look at factors such as whether they’re supplying materials for implants or other vital aspects which can directly impact patient health. There should always be a connection between the supplier and the particular product being bought; suppliers are only approved for particular materials. Criticality can be measured in terms of either product or business-critical.
With a risk-based QMS, companies need to examine their suppliers and determine what “critical” and “noncritical” means. The wrong materials can have huge ramifications from both a patient and manufacturing standpoint. This is risk-based supplier management.
A key part of a risk-based approach is that you should monitor and evaluate suppliers with some frequency. If there are any issues with the things you are buying from the supplier, you should take action to mitigate your risk and respond appropriately. This might include actions such as finding a new supplier or issuing a current supplier with a formal corrective action. The overall idea is that you apply a process to assess risk to your supplier management process.
Here is a relevant piece for any outsourced processes:
“When the organization chooses to outsource any process that affects product conformity to requirements, it shall monitor and ensure control over such processes. The organization shall retain responsibility of conformity to this International Standard and to customer and applicable regulatory requirements for outsourced processes. The controls shall be proportionate to the risk involved and the ability of the external party to meet the requirements in accordance with 7.4. The controls shall include written quality agreements.”
Non-conformance is closely associated with manufacturing. You’ll need to determine the disposition of the error by asking yourself the following questions: What do I do with this? Can I use it as it is? Do I need to rework it? Do I scrap it altogether?
The decision about what to do is a risk-based approach to non-conformance. The expectation for being risk-based is that you must factor in the risk of each decision on the manufacturing process and/or the patient.
Historically companies don’t go to this level. They have a checkbox on a form — “was risk impacted by this?” — with a yes/no response. There’s no connection made with the risk documentation and it operates as a siloed function. The aim of new requirements is that this is no longer the case. A holistic approach to risk management helps to avoid silos and improve transparency across the whole operation.
You can find the revised information under sections 8.3 and 8.3.4 of ISO 13485:2016. They don’t mention the word risk, but they do use the term “adverse effect”, which can be understood to mean risk. The bottom line is that they want you taking risk into account whenever you have to perform rework due to non-conformance.
Similarly to the solution for non-conformances, companies often use a simple checkbox for this. They really should be shifting to a product-centric interpretation of risk. What does it mean for the product or product family? Have you defined probability and severity? Was it already documented in your risk management file? Was the risk already defined correctly or does it need updating?
The real risk when you launch your product may well be higher than you first thought so it’s important to keep assessing based on the feedback that you receive and any other incidents.
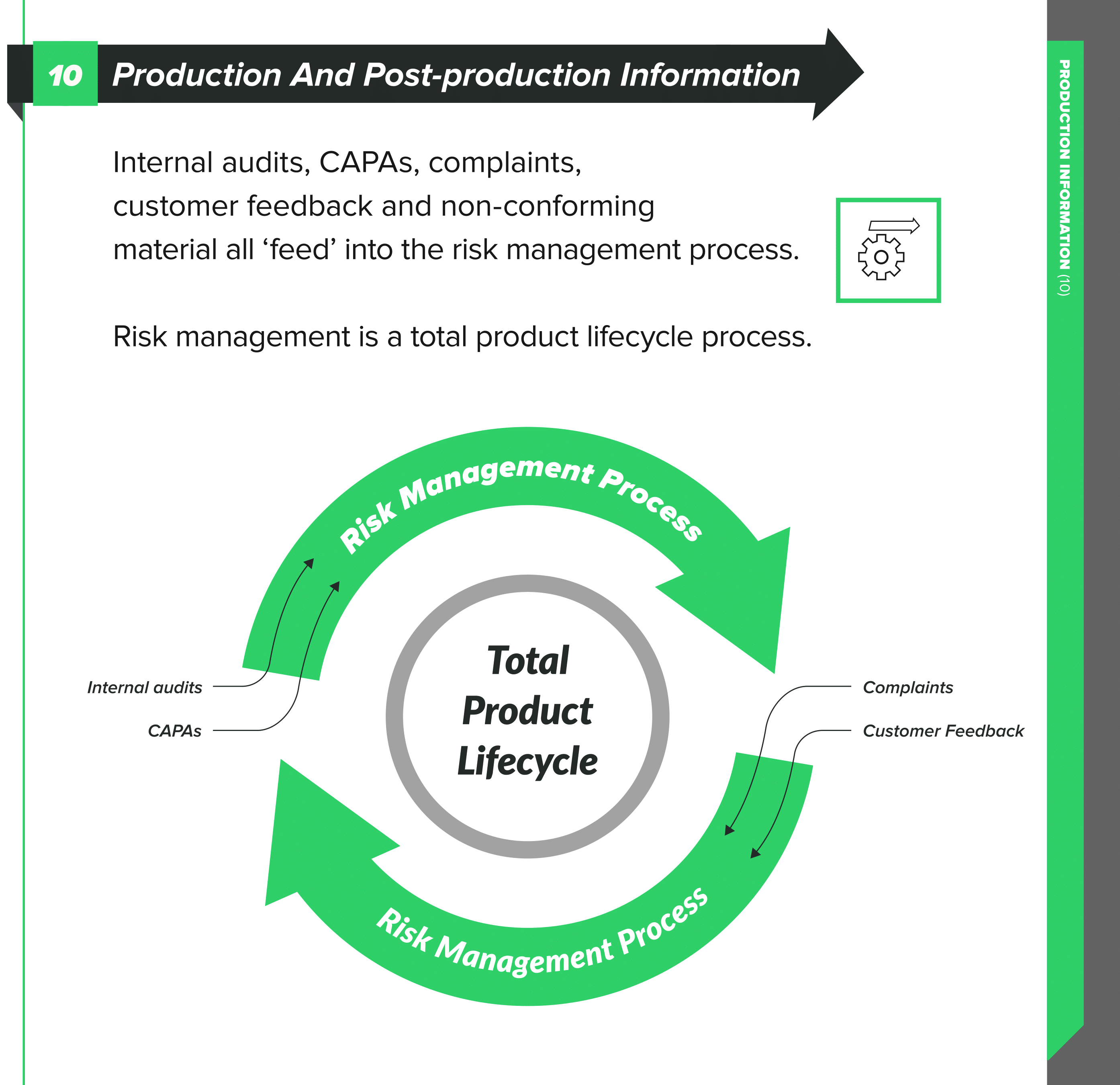
Have a risk management file for the product so that you can go back to a knowledge base for the product when complaints happen. As we have discussed in previous articles, risk management is an activity spanning the full product lifecycle, not just something you do during design and production.
Within ISO 13485:2016, check out sections 8.2, 8.2.1, and 8.2.2 for their advice on feedback and complaint handling. You need to have documented procedures for the feedback process and include provisions to gather data from the production and post-production phases.
The feedback that you gather becomes potential inputs into risk management. You need to analyze it from a risk perspective and evaluate with a view to the safety of the patient and the performance of the device as intended. Your feedback might provide you with cause to make changes to either the design or something in your processes, but again, you need to analyze the risk of these changes.
People often assess risk within the context of CAPA. This is another example of risk handled in the same siloed way that we see in each of the above areas, rather than holistically across the product.
CAPA is for systemic issues. If it is product or manufacturing-related, it’s virtually guaranteed that risk is negatively impacted. You need to go back and update the risk management file and doing so should be an action point as part of the CAPA.
An example of integrating risk management with CAPA is that your CAPA records could include extended data to show the impact and likelihood of such an event. This can help with sorting and filtering the CAPA records to create some kind of priority view.
A risk-based QMS means applying a process to assess risk to each of these processes.
A QMS is our architecture for demonstrating all the things we do to comply with regulations. Those actions tend to be good business practices, too. If you think of risk as a hierarchy, we’re looking for those underlying QMS processes to be as risk-averse as possible.
The new ISO 13485:2016 standard puts more emphasis on risk management before product realization. This means organizations need to show how they are making risk-based rather than rule-based decisions to comply with the regulations. Doing so can bring the benefit of better allocation of resources across your business.
Still using a manual or paper-based approach to manage your design controls or quality processes? Click here to learn more about how greenlight.guru's modern eQMS software platform exclusively for medical device companies is helping device makers all over the globe in more than 50 countries get safer products to market faster with less risk while ensuring regulatory compliance.